БЧЛб»Ҝө°°ЧҪMЙъРЕ·ЦОц
LeveneәНAlsbergФЪ1906ДкКЧҙО°l¬FБЛө°°ЧөДБЧЛб»ҜЈ¬Я@ӮҖPTMФЪҪь30Дкәуұ»¶ЁО»өҪө°°ЧөДҪz°ұЛбҡҲ»щЙПЈЁИзҲD1Ј©ЎЈУЙУЪЛДӮҖоIУтөДЖҪРР°lХ№Јә(i)¶юҫSДэДzлҠУҫ(2D-PAGE)Ј»(ii)Щ|ЧV·Ҫ·ЁЈ»(iii)ө°°Ч”ө“юҺмЈ»(iv)ЙъОпРЕПўҢW№ӨҫЯЈ¬ө°°ЧЩ|ҪMҢWСРҫҝФЪ1990ДкҙъЦРЖЪй_Кј[1]ЎЈ
ЧоіЈТҠөДө°°ЧЩ|БЧЛб»Ҝ°ьАЁЕcҪz°ұЛбЎўМK°ұЛбәНАТ°ұЛбөДБu»щӮИжңРОіЙБЧЛбхҘжIЎЈғЙ·NЮЧҝ№ГёПөҪyЈЁјӨГёәНБЧЛбГёЈ©·Ц„eҙЯ»Ҝө°°ЧЩ|БЧЛб»ҜәНИҘБЧЛб»Ҝ[2]ЎЈБЧЛб»Ҝұ»·ЦһйЛДоҗЈәO-phosphorylation(pSer, pThr, pTyr), N-phosphorylation(pHis, pArg, pLys), phosphoanhydride(pAsp, pGlu) and phosphorothioate (pCys)ЈЁТҠҲD2Ј©[3]ЎЈ
ө°°ЧЩ|БЧЛб»ҜКЗФS¶ајҡ°ыЯ^іМЦРөД»щұҫХ{№қҷCЦЖЈ¬БЧЛб»ҜөД®җіЈ”_„УЕcёч·NИЛоҗјІІЎУРкPЎЈТтҙЛЈ¬ДЬүтҢҰБЧЛб»Ҝө°°ЧЩ|ҪMЯMРРИ«ПөҪy¶ЁБҝ·ЦОцҢўһйҪТКҫёРЕdИӨөДјІІЎөДРВРЕМ–НЁВ·ЎўЛҺОп°РьcәНЙъОпҳЛЦҫОпМṩҸҠҙуөДНЖ„УБҰЎЈ

ҲD1 ФзЖЪБЧЛб»ҜСРҫҝәҶК·[1]
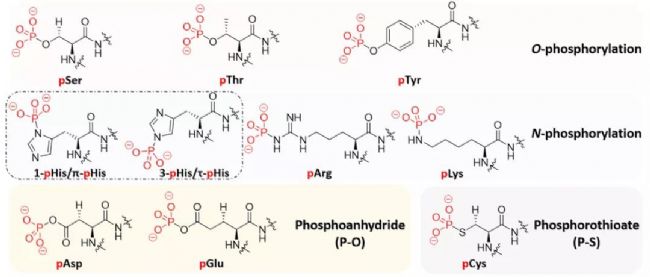
ҲD2 БЧЛб»ҜРЮп—О»ьcКҫТвҲD[3]
1.нf¶чҲD
І»Н¬МҺАнҪMЦ®йgөД№ІРФәНІо®җНЁіЈҝЙТФУГнf¶чҲDХ№Кҫ[4]ЎЈBioVenn1ДЬұн¬FіцІ»Н¬ҪMөДФӘЛШ”өБҝТФј°ПаН¬ьcәНІ»Н¬ьcЈ¬Н¬•rХ№¬F”ө“юјҜөДҙуРЎЎЈ
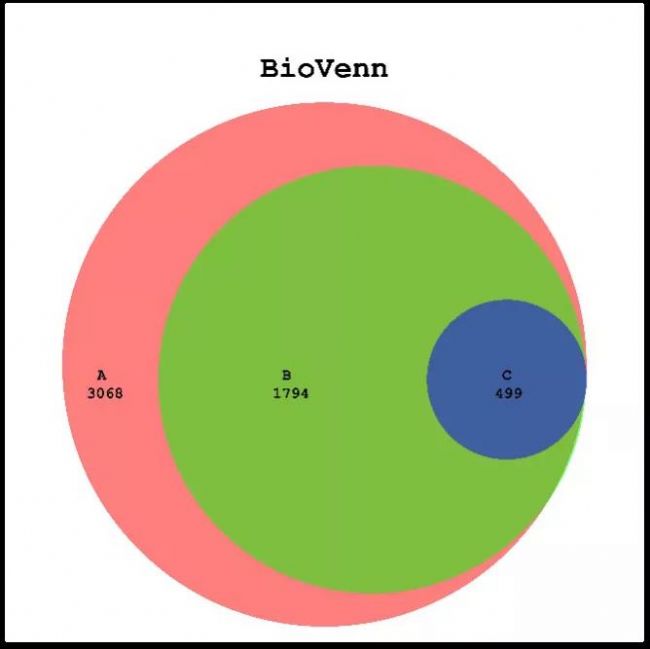
ҲD3 BioVenn-1
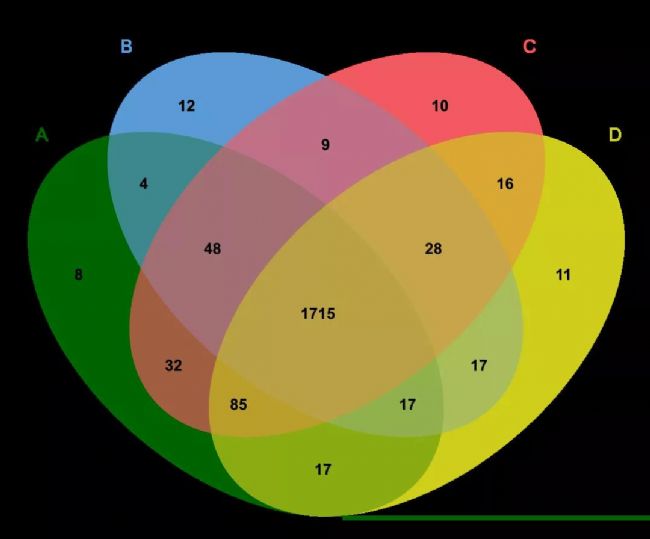
ҲD4 BioVenn-1
2.»рЙҪҲD
ФЪCHO-K1јҡ°ыЯm‘Әҹo№И°ұхЈ°·ЕарB»щөДЙъйLөДҢҚтһЦРЈ¬УГ»рЙҪҲDҒнұнКҫҪMйgІо®җұнЯ_ө°°ЧөДұнЯ_БҝЧғ»ҜЈ¬й“ЦөФOЦГһйfold change < 1.5әНp value < 0.05[5]ЈЁИзVolcano Plot-1Ј©ЎЈІўФЪҲDЦРҳЛЧўTOP10Іо®җұнЯ_ө°°ЧЎЈФЪҢҚтһЦРЈ¬ұнЯ_БҝёЯЗТп@ЦшРФёЯөДІо®җө°°ЧКЗИЭТЧұ»кPЧўөДЈ¬Іўұ»ЯxһйДҝҳЛө°°ЧЎЈЯ@ТІДЬҪөөНәуАmөДҢҚтһлy¶ИЎЈ
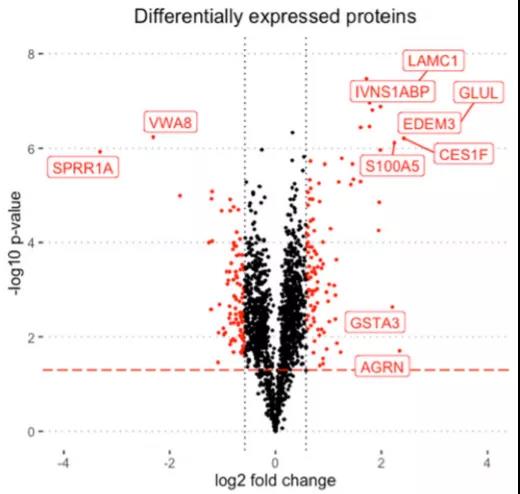
ҲD5 Volcano Plot-1[5]
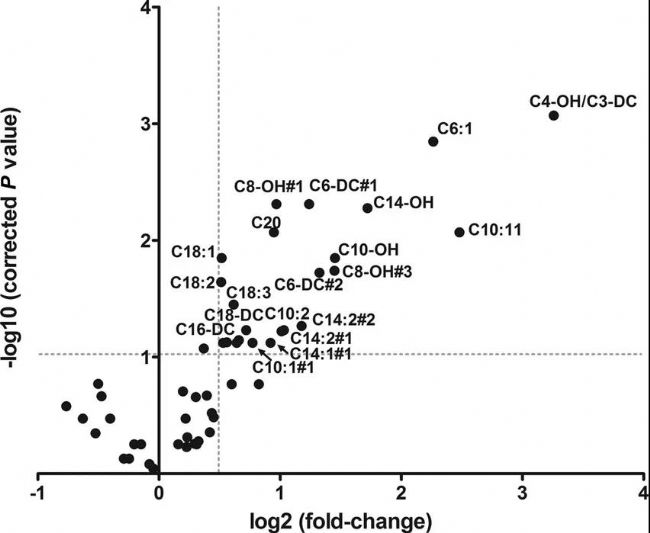
ҲD6 Volcano Plot-2[6]
3.KEGG Pathway·ЦОц
БЧЛб»ҜЛ®ЖҪ•ғТ№ТҺВЙІЁ„УөДёОЕKө°°Чп@Цшё»јҜФЪМШ¶ЁөДҙъЦxНЁВ·ЙПЈ¬ИзТИҚuЛШПакPҙъЦxНҫҸҪЎўјҡ°ыЧФКЙәН•ғТ№№қВЙПакPөДҙъЦxНҫҸҪЈЁИзҲDKEGG PATHWAY1-2Ј©Ј¬ҲDЦРҷMЧшҳЛһйё»јҜіМ¶ИRich factorЦөЈ¬ҝvЧшҳЛұнКҫіCХэәуөДP-valueЦөөДҙуРЎЈ¬ұнГч·ӯЧgәуРЮп—ФЪ•ғТ№№қВЙХ{ҝШЦРЖрЦшкPжIЧчУГЎЈ
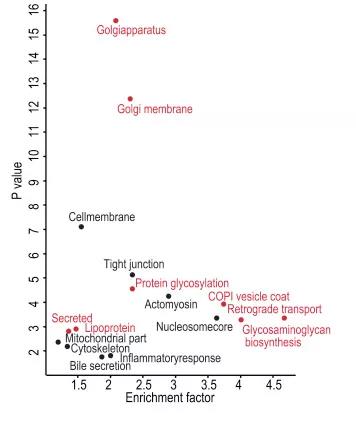
ҲD7 KEGG pathway-1[7]
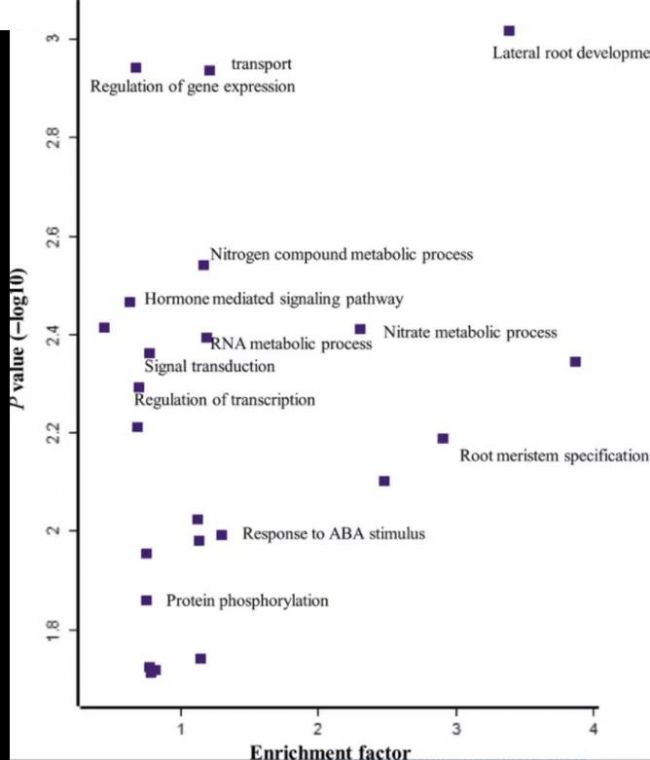
ҲD8 KEGG pathway-2[8]
4.GO·ЦОц
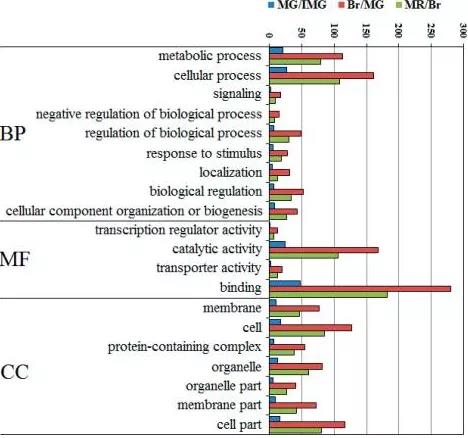
GO·ЦОцЦјФЪ°lҫтЕc»щТтІо®җұнЯ_¬FПукPВ“өДҶОӮҖМШХч»щТт№ҰДЬоҗ»т¶аӮҖМШХч№ҰДЬоҗөДҪMәПЎЈПВГжОТӮғТФАұҪ·өДИэӮҖІ»Н¬іЙйLлA¶ОһйАэЈ¬ТФЦұ·ҪҲDөДРОКҪХ№КҫБЛGO·ЦОцөДҪY№ыЎЈҲDЦРҷMЧшҳЛһйУіЙдІо®җұнЯ_ө°°ЧөДӮҖ”өЈ¬ҝvЧшҳЛһйGO TermЎЈ
ҲD9 GO-1[9]
ҲD10 GO-2[10]

ҲD11 GO-3[8]
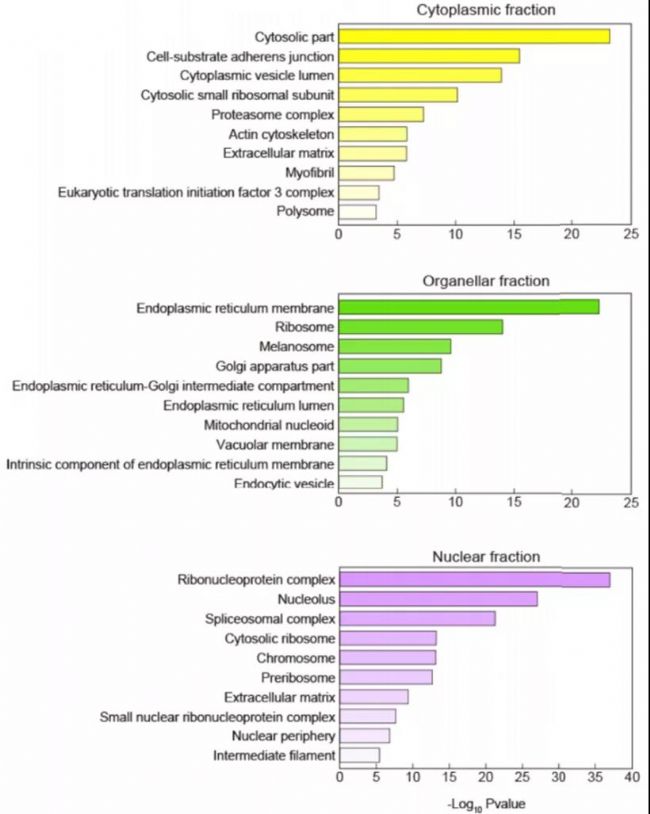
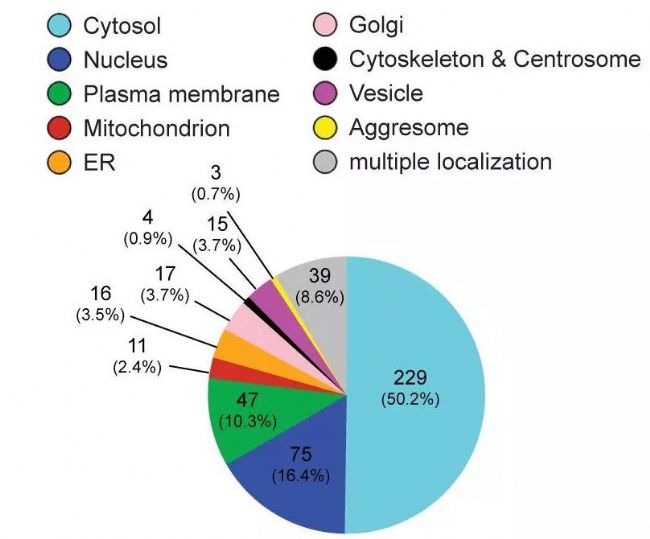
5.ҒҶјҡ°ы¶ЁО»
ө°°ЧЩ|ұШнҡЮDЯ\өҪЖд‘ӘФЪөДҒҶјҡ°ыҪYҳӢЙПІЕДЬРРК№Жд№ҰДЬЈ¬·с„tҫН•юіц¬FҷCуw№ҰДЬОЙҒyЈ¬®aЙъёч·NјІІЎЎЈө°°ЧЩ|өДО»ЦГКЗө°°ЧЩ|ЧоЦШТӘөДҢЩРФЦ®Т»Ј¬УРЦъУЪҙ_¶Ёө°°ЧЩ|№ҰДЬЈ¬ҪТКҫ·ЦЧУҪ»»ҘҷCАнЎўАнҪвҸНлsЙъАнЯ^іМәНй_°lЛҺОп°РҳЛөИ·ҪГжөДСРҫҝЎЈ
ҲD12 Subcellular Location[11]
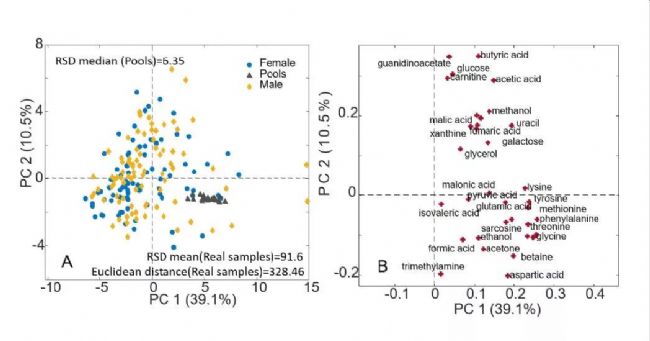
6.PCA·ЦОц
ЦчіЙ·Ц·ЦОц (PCA, principal component analysis)КЗТ»·N”өҢWҪөҫS·Ҫ·Ё, АыУГХэҪ»Чғ“Q (orthogonal transformation)°СТ»ПөБРҝЙДЬҫҖРФПакPөДЧғБҝЮD“QһйТ»ҪMҫҖРФІ»ПакPөДРВЧғБҝЈЁТІ·QһйЦчіЙ·ЦЈ©Ј¬ҸД¶шАыУГРВЧғБҝФЪёьРЎөДҫS¶ИПВХ№КҫСРҫҝҢҰПуөДМШХчЎЈУГЙЩ”өҺЧӮҖЦчіЙ·ЦөДЧғ»ҜҒнҪьЛЖҙъМжФӯҒн¶аӮҖЧғБҝөДЧғ»ҜЎЈНЁЯ^ЦчіЙ·Ц·ЦОцЈ¬ҝЙТФ·ҙУіІ»Н¬ҳУұҫйgөДкPПөЈ»ҢҰУЪҙуБҝҳУұҫЈ¬НЁЯ^лxИәьcЈ¬ЕР”аКЗ·сҙжФЪлxЙўҳУұҫЈ¬ФЪЯMТ»ІҪөД·ЦОцЦРЯMРРМЮіэЎЈ
 ҲD13 PCA[12]
ҲD13 PCA[12]
7.PPI·ЦОц
ө°°ЧЩ|-ө°°ЧЩ|Па»ҘЧчУГҫWҪj·ЦОц(Protein-Protein Interaction Network Analysis, PPI Network Analysis)Ј¬КЗө°°ЧЩ|ҪMҢWөДЦШТӘСРҫҝғИИЭЦ®Т»ЎЈө°°ЧЩ|ФЪРРК№ЙъОп№ҰДЬ•rНЁЯ^РОіЙPPIҫWҪjТФҫSіЦ•rйgәНҝХйgЙПөД…fХ{Т»ЦВЈ¬ҳӢҪЁІо®җұнЯ_ө°°ЧөДПа»ҘЧчУГҫWҪjЈ¬ҝЙТФҸДө°°ЧЩ|ҪMҢУГж°l¬FІо®җұнЯ_ө°°ЧөДЧғ»ҜЪ…„ЭЈ¬ЯMТ»ІҪҺНЦъОТӮғҢӨХТІо®җұнЯ_ө°°ЧЦРөДкPжI№қьcЎЈ
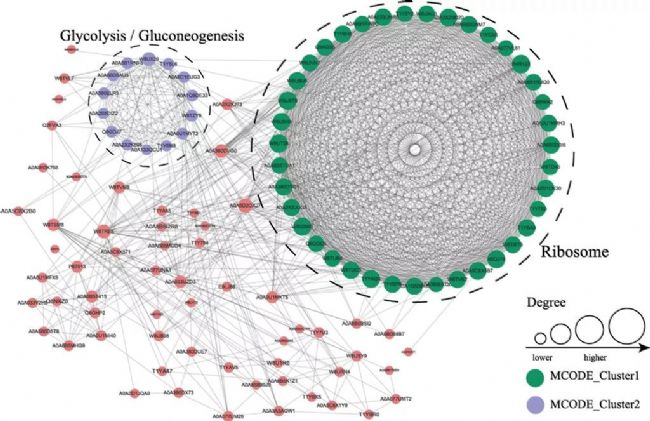

8.Motif·ЦОц
ГёҢҰМШ¶ЁөЧОпөДІҝ·ЦЙъ»ҜЖ«әГҝЙДЬКЗУЙРЮп—О»ьcЦЬҮъөДҡҲ»щӣQ¶ЁөДЈ¬Я@·Nө°°Ч»т¶алДРтБРРОіЙөДМШ¶ЁҡҲ»щДЈКҪ·Qһй»щРтЈЁMotifЈ©ФЪө°°ЧөДН¬ФҙРтБРЦРЈ¬І»Н¬О»ьcөДұЈКШіМ¶ИКЗІ»Т»ҳУөДЈ¬Т»°гҒнХfЈ¬ҢҰө°°ЧЩ|№ҰДЬәНҪYҳӢУ°н‘ұИЭ^ҙуөДО»ьc•юұИЭ^ұЈКШЈ¬ЖдЛьО»ьc„tІ»КЗәЬұЈКШЎЈЯ@Р©ұЈКШөДО»ьcҫН·Qһй“ДЈуw(motif)”ЎЈҝЙТФёщ“юө°°ЧЩ|РтБРМШХчЈЁұИИз»щУЪө°°ЧЩ|»щРтЈ©ЯMРР№ҰДЬоAңyЎЈ
ҲD16 motif-1[15]

ҲD17 motif-2
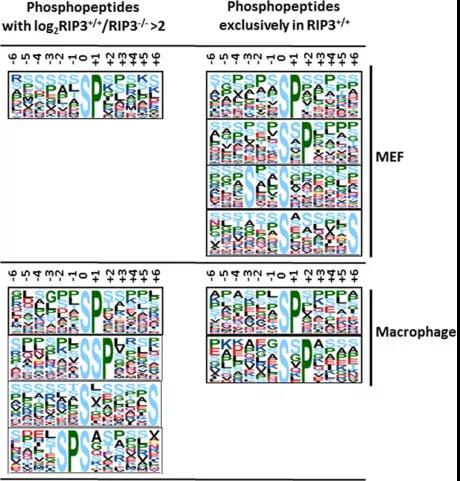
9.јӨГё(kinase)оAңy·ЦОц
ө°°ЧјӨГёЈЁprotein kinasesЈ¬әҶ·QPKЈ©ЎЈҙЯ»Ҝө°°ЧЩ|БЧЛб»ҜЯ^іМөДГёЎЈө°°ЧЩ|өДБЧЛб»ҜЯ^іМКЗЙсҪӣРЕПўФЪјҡ°ығИӮчЯfөДЧоәуӯh№қЈ¬Ң§ЦВлxЧУНЁөАө°°Чј°НЁөАйTөД о‘BЧғ»ҜЎЈТтҙЛЈ¬јӨГёөДЧғ»ҜЕc°©°YәНјІІЎГЬЗРПакPЎЈФЪБЧЛб»Ҝө°°ЧҪM·ЦОцЦРЈ¬НЁіЈАыУГmotifҒноAңyјӨГёЈ¬ҪТКҫ…ўЕc·ЦЧУЯ^іМөД…ўЕcХЯЎЈіЈУГоAңyјӨГёҫWХҫЈәhttp://hprd.org/PhosphoMotif_finderЎЈ
…ўҝјОД«I
[1] Dupree, E.J., et al., A Critical Review of Bottom-Up Proteomics: The Good, the Bad, and the Future of this Field. Proteomes, 2020. 8(3).
[2] Kjeldsen, F., et al., On studying protein phosphorylation patterns using bottom-up LC-MS/MS: the case of human alpha-casein. Analyst, 2007. 132(8): p. 768-76.
[3] Huang, B., et al., NMR-based investigation into protein phosphorylation. Int J Biol Macromol, 2020. 145: p. 53-63.
[4] Panizza, E., et al., Isoelectric point-based fractionation by HiRIEF coupled to LC-MS allows for in-depth quantitative analysis of the phosphoproteome. Scientific Reports, 2017. 7(1): p. 4513.
[5] Kaushik, P., et al., LC-MS/MS-based quantitative proteomic and phosphoproteomic analysis of CHO-K1 cells adapted to growth in glutamine-free media. Biotechnology Letters, 2020. 42(12): p. 2523-2536.
[6] Ruiz M, Labarthe F, Fortier A, Bouchard B, Thompson Legault J, Bolduc V, Rigal O, Chen J, Ducharme A, Crawford PA, Tardif JC, Des Rosiers C. Circulating acylcarnitine profile in human heart failure: a surrogate of fatty acid metabolic dysregulation in mitochondria and beyond. Am J Physiol Heart Circ Physiol. 2017 Oct 1, 313(4).
[7] Krahmer N, Najafi B, Schueder F, et al. Uhlenhaut NH, Walther TC, Jungmann R, Zeigerer A, Borner GHH, Mann M. Organellar Proteomics and Phospho-Proteomics Reveal Subcellular Reorganization in Diet-Induced Hepatic Steatosis. Dev Cell. 2018 Oct 22;47(2): 205-221.
[8] Chen Y, Hoehenwarter W. Rapid and reproducible phosphopeptide enrichment by tandem metal oxide affinity chromatography: application to boron deficiency induced phosphoproteomics. Plant J. 2019 Apr;98(2):370-384.
[9] Liu Z, Lv J, Liu Y, Wang J, et al . Comprehensive Phosphoproteomic Analysis of Pepper Fruit Development Provides Insight into Plant Signaling Transduction. Int J Mol Sci. 2020 Mar 13;21(6).
[10] Masuda T, Sugiyama N, Tomita M, Ohtsuki S, Ishihama Y. Mass Spectrometry-Compatible Subcellular Fractionation for Proteomics. J Proteome Res. 2020 Jan 3;19(1):75-84.
[11] Zhang H, Cao X, Tang M, Zhong G, Si Y, Li H, Zhu F, Liao Q, Li L, Zhao J, Feng J, Li S, Wang C, Kaulich M, Wang F, Chen L, et al. A subcellular map of the human kinome. Elife. 2021 May 14;10:e64943.
[12] Cui Mengni,Trimigno Alessia,Aru Violetta,Rasmussen Morten A,Khakimov Bekzod,Engelsen Søren Balling. Influence of Age, Sex, and Diet on the Human Fecal Metabolome Investigated by 1H NMR Spectroscopy.[J]. Journal of proteome research,2021.
[13] Shi Y , Zhu J , Xu Y , et al. Malonyl-proteome profiles of Staphylococcus aureus reveal lysine malonylation modification in enzymes involved in energy metabolism[J]. Proteome Science, 2021,19(1).
[14] Li H , Y Zhang, Zhang J , et al. A quantitative proteomics analysis for small molecule Stemazole’s effect on human neural stem cells[J]. Proteome Science, 2020, 18(1).
[15] Wu X, Tian L, Li J, et al. Investigation of receptor interacting protein (RIP3)-dependent protein phosphorylation by quantitative phosphoproteomics. Mol Cell Proteomics. 2012 Dec;11(12):1640-51.
[16] Grosstessner-Hain K, Hegemann B, Novatchkova M, et al. Quantitative phospho-proteomics to investigate the polo-like kinase 1-dependent phospho-proteome. Mol Cell Proteomics. 2011, 10(11).

- Target-BSјјРgҪТКҫМЗДтІЎТэ°lІӘЖр№ҰДЬХПөKөДDNAјЧ»щ»ҜХ{ҝШҷCЦЖ
- әПіЙЙъОпҢWөЧұPјҡ°ыЦ®®…іаҪНДёіЈУГұнЯ_Эdуwј°»щТтёДФмјјРg
- Йо¶ИҪвОцCRISPRОДҺмәYЯxБчіМј°‘ӘУГ°ёАэ
- ҝЙңШәН—lјюПВёЯ·ЦұжВКҷzңym6AРЮп—өДРВm6AңyРт·Ҫ·Ёй_°lСРҫҝ
- AIИЛ№ӨЦЗДЬФЪө°°ЧЩ|ҪYҳӢоAңyЎў№ҰДЬоAңyј°ФOУӢЦРөДЧчУГ
- m5C MeRIP-seqөИҪТКҫm5CРЮп—ФЪ°©°YДНЛҺЦРөДкPжIХ{ҝШҷCЦЖЦРөД‘ӘУГ
- ChIPјјРgҪТКҫNURR1ФЪЗ°БРПЩ°©ҸД»щТтЮDдӣөҪД[БцЯMХ№ЦРөДХ{ҝШҷCЦЖ
- ғЙ·NЮDдӣТтЧУФЪҪйҢ§·¬ЗС№ыҢҚіЙКмөДұнУ^ЯzӮчХ{ҝШЦР°l“]өДкPжIЧчУГ
- 2025І®әАЙъОпҙәјҫИ«ҮшСІЦvй_ҶўЈ¬ФзшBҲуГы“ҢХјПИҷC
- ИAҙуЦЗФмёұҝӮІГЦРҮш…^ҝӮҪӣАнЕнҡgҡgТ»РРөҪФLІ®әАЙъОп
- І®әА¶ӯКВйLіцПҜҢЈҫ«МШРВЦРРЎЖуҳI°lХ№үСҙуХ“үҜІў°lСФ
- І®әАЙъОплp11ҝсҡgМШ»Э»о„УҒнТuЈ¬ПЮ•rПЮБҝ“Ң
- вщГАНЁөВНЖіціЙұҫғrуwтһҶОјҡ°ыңyРтЈЁFFPE»тИ«СӘЈ©
- І®әАЙъОп2024ПөБРЕаУ–°аЈЁкғОчХҫЈ©ҲуГый_Ҷў
- І®әАҶОјҡ°ыFlexДкЦРҫЮ»ЭЈ¬1.1W/ҳУұҫ»о„УғrјҙҢўҪШЦ№
- Л{ҫ°ҝЖРЕDAP-seqјјРgПакPОДХВ6ЯB°lЈ¬ҝӮIF 95.2