從SWATH到MRM:一種新型高覆蓋度靶向代謝組學技術的介紹

代謝組學文獻分享—摘要
生物樣本代謝組存在高度復雜性,對大規模定量研究這些代謝物帶來極大的挑戰,嚴重限制了代謝組學在生物標志物發現研究中的運用。靶向代謝組學研究主要運用MRM技術,具有極高的定量能力,但卻受制于較為有限的覆蓋度。為了解決這一挑戰,作者開發了一項新的技術策略——SWATHtoMRM,巧妙的運用了SWATH技術的高覆蓋度特點來開發高覆蓋度的靶向代謝組學方法。
首先,運用SWATH技術對一個生物混樣進行非靶向代謝譜分析,以獲取其中“所有”代謝物的二級譜圖 (MS²);然后從中提取MRM離子對信息,用于構建高達1000-2000代謝物覆蓋度的靶向代謝組學分析;隨后,作者又展示了這種方法在定量代謝組學中優勢,例如:覆蓋度、重復性、靈敏度和動態范圍等。最后,作者運用SWATHtoMRM技術尋找colorectal cancer (CRC)診斷標志物,單針進樣便可完成多達1303種代謝物MRM檢測。從CRC病人組織中發現并驗證了20種潛在診斷標志物。其中17種標志代謝物在CRC病人血漿中被進一步驗證與腫瘤切除相關,有著極大的CRC術后預后標志物潛力。
SWATHtoMRM技術為我們提供了一種全新的途徑來開發高覆蓋度的靶向代謝組學方法,將促進靶向代謝組學在疾病標志物發現研究中的應用。
代謝組學文獻分享—背景
代謝組學旨在系統性定量檢測所有代謝物的動態變化,為疾病表型提供一個綜合的特征描述,以便于疾病標志物的發現。代謝物具有廣泛的結構多樣性和懸殊的濃度范圍(>109)。因此,代謝組學研究要求分析技術具有廣泛覆蓋度、高靈敏性和特異性以及寬動態范圍。
液質聯用(LC-MS)技術是較為流行的代謝譜分析技術之一,包括靶向和非靶向兩種方式。非靶向代謝組學多運用高分辨質譜(TOF/Orbitrap)來檢測盡量多的代謝物,具有很寬的覆蓋度,但是受限于動態范圍、定量精度以及靈敏度等。相反,基于LC-MS的靶向代謝組學技術主要使用MRM技術,能夠精確定量一定數量(100-300)的已知代謝物。
靶向代謝組學技術作為代謝物定量的金標準,具有較高的靈敏度、動態范圍和較好的重現性。然而,較低的代謝物覆蓋度成為了靶向代謝組學的主要運用瓶頸,因此,開發一種具有高覆蓋度的靶向方法來準確定量在非靶向代謝組學中檢測到的已知/未知代謝物至關重要。
針對MRM覆蓋度低這一瓶頸,近期已有多篇文章著手開發高覆蓋度的靶向代謝組學技術。 但這些技術往往需要很長的機時(20-30針, 有的甚至需要80針)來獲取代謝物的二級譜圖,嚴重限制了該技術的通量;使用常規的DDA采集模式來獲取代謝物二級譜圖也是這些方法中的一種,但由于DDA模式本身的隨機性和偏好性,使得這種采集方式重復性低,覆蓋度也偏低。不同于DDA采集模式,DIA/SWATH模式能夠高效、高覆蓋度獲取“所有”代謝物的二級譜圖,在近幾年被不斷開發用于非靶向代謝組學研究。
盡管SWATH技術能夠同時采集所有代謝物的二級碎片離子信息,但是在復雜生物樣品中,它的定量動態范圍和重復性都受到了很大的限制。正是由于任何單一技術都不是廣泛適用的,本文作者集各家之所長,將SWATH技術強大的定性能力與MRM技術的精確定量能力完美結合,開發出了SWATHtoMRM技術。
代謝組學文獻分享—方法
LC-MS方法
為了保證SWATH采集到的代謝物二級碎片離子信息與MRM中所使用的Q1/Q3離子對信息相對應,需要保證液相條件(RT)完全一致,并盡量保證SWATH采集所使用的Q-TOF質譜儀(TripleTOF 6600)與MRM采集所使用的QQQ質譜儀(Agilent 6495 QQQ)具有相對一致的各項參數(CE,DP等)。
數據處理方法
SWATHtoMRM流程
SWATHtoMRM技術的分析流程已整理成R包,用于從SWATH數據中大規模提取MRM離子對信息,該R包可以在(http://www.zhulab.cn/software.php)下載安裝。整個流程分為兩步(圖1):
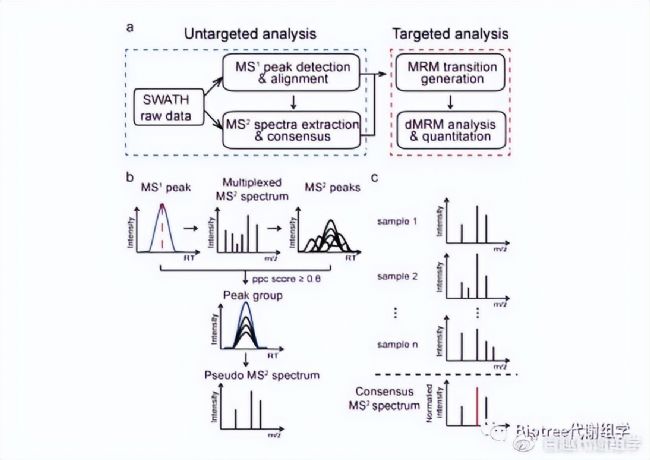
圖1. (a) SWATHtoMRM 技術流程概覽。 (b) 從SWATH數據中提取虛擬MS²圖譜。 (c) 將來自多個樣品的MS²譜圖合并為consensus 譜圖,保留出現頻率超過50%的碎片離子,并用于生成MRM離子對。
1.非靶向分析SWATH數據
a) 使用XCMS包中的CentWave算法實現色譜峰(LC peak)識別,然后用CAMERA進行質譜峰(MS peak)注釋,并去除同位素峰和小于1000的弱信號峰。對于多個數據文件,使用OBI-Warp算法進行峰對齊。→MS¹ LC peak 。
b) 針對每一個MS¹peak,從峰頂位置找到對應RT的混合MS²譜圖,并過濾掉小于200的弱信號值、同位素離子以及大于母離子m/z的碎片離子。提取保留的碎片離子色譜峰(EIC)。→MS² LC peak 。
c) 通過計算每一個碎片離子EIC與母離子EIC之間的相關性得分(PPC score),將得分大于0.8的碎片離子EIC和對應母離子EIC歸為一組,得到peak group(一個母離子EIC +多個碎片離子EIC),然后對每個peak group生成一張虛擬二級譜圖。→ Pseudo MS² spectrum。
d) 將來自多個樣品的相同MS¹ peak的二級譜圖合并為一張譜圖(consensus MS²)。合并原則為:保留出現頻率超過50%的碎片離子,合并后的m/z為mean(m/z),intensity %為 mean(intensity %)。→ Consensus MS² spectrum。
2.生成MRM離子對信息
a) 采用3個原則評估consensus MS²中每一個碎片離子:(1)m/z(product ion)使用Agilent MassHunter構建schedule MRM方法,最小dwell 時間 5ms,cycle time為990ms。
代謝組學文獻分享—結果與討論
SWATHtoMRM 流程評估
首先,作者測試了SWATHtoMRM技術在多種生物樣品,例如,人類尿液、結腸直腸組織以及Jurkat細胞中的適用性。
以人尿液樣品為例,3554個MS¹ feature在SWATH數據中被檢測到,其中950個同位素峰和弱信號峰被初步過濾。剩余的2604個代謝物峰中,2091 (80.3%) 個代謝物具有合適的MRM離子對信息,其他代謝物峰由于不滿足PPC > 0.8這一條件被進一步剔除。通過手動分析2091個MRM離子對結果發現,多達1614 (77.2%) 個代謝物成功在尿液樣品中檢測到(圖2a)。
類似的,在其他類型樣品中得到了接近的檢出率(76 - 80%)。 本次實驗中僅采用了正離子模式,但負離子模式也是完全適用的。
圖2b詳細演示了通過SWATHtoMRM構建MRM離子對的過程。 以taurine這個代謝物(m/z = 126.0212 Da,RT= 518 s)為例:首先,包含多個代謝物碎片離子的多重二級譜圖被提取出來。然后根據與母離子EIC之間的相關性PPC,保留得分大于0.8 的碎片離子。隨后,將來自多個樣品的同一代謝物二級譜圖合并,得到一張包含13個碎片離子的consensus二級譜圖。最后,從中選擇響應強度最高的碎片離子(m/z = 44.0496 Da)構建taurine的MRM離子對(Q1/Q3, 126.0/44.0)。

圖2. (a) 使用人尿液樣品大規模生成MRM離子對。(b) 代謝物taurine的離子對生成過程。 (c) 從不同類型生物樣品(尿液,組織和細胞)中檢測到的MRM離子對統計信息。
SWATHtoMRM技術的覆蓋度評估
為了證實這一技術具有廣泛的覆蓋度,作者將SWATHtoMRM方法與DDA方法做了系統的比較。使用相同的尿液樣品,相同的儀器參數,連續采集SWATH和DDA數據。后續的數據分析也采用相似的參數自動化進行,以較大的降低主觀偏好性。從相同的尿液樣品中,分別檢測出2604(SWATH)和2149(DDA)個feature(圖3a, 3b)。比較兩者的二級譜圖覆蓋度,SWATH(2105, 80.8%)顯著高于DDA(1174, 54.6%)。二級覆蓋度在兩者共享feature中,SWATH(84.9%)也顯著高于DDA(61.1%)。
SWATH與DDA技術分別成功構建2091和1163個MRM離子對,其中852個是共享離子對(圖3c)。在這852個共享離子對中,分別有87.2%(SWATH)和88.7%(DDA)的檢出率,兩種方法具有接近的檢出率,說明SWATH技術生成的MRM離子對信息是可靠的(圖3d)。 但是從總檢出數上來看,SWATH技術比DDA多出66%的代謝物。簡言之,SWATH技術具有與DDA相似的數據質量,同時又顯著超越了DDA的檢測數量。
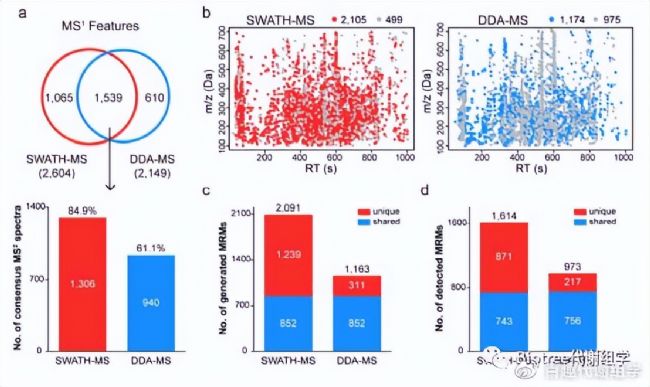
圖3. 從SWATH數據中生成的MRM離子對具有很廣的覆蓋度。(a) 韋恩圖展示了SWATH與DDA技術檢測到的共享和特有feature。(b) SWATH與DDA數據中MS¹和MS²的分布圖。紅/藍=有MS²,灰色=無MS²。(c) 兩種方法構建的MRM離子對數目比較。(d) 兩種方法檢測到的代謝物數目比較。
SWATHtoMRM定量性能評估
為了評估SWATHtoMRM技術的定量性能,作者對比了SWATHtoMRM,SWATH-MS¹以及SWATH-MS² 3種技術間的靈敏度、動態范圍和重復性。首先,梯度稀釋尿液樣品,分別進行SWATHtoMRM和SWATH采集,然后隨機選擇629個檢測到的代謝物進行比較。
1.靈敏度:比較不同稀釋梯度中代謝物檢出數來評估,結果SWATHtoMRM > SWATH-MS¹= SWATH-MS²(圖4a)。將這一比較細化到不同豐度范圍代謝物中,同樣是SWATHtoMRM優勝,尤其是在低豐度代謝物中(圖4b)。
2.動態范圍:比較不同稀釋梯度中,629個代謝物的定量線性范圍(R²)來評估,結果SWATHtoMRM > SWATH-MS¹> SWATH-MS²,SWATHtoMRM數據中R²> 0.8的代謝物超過了80%,而SWATH-MS¹和SWATH-MS²則不到60%(圖4c)。
3.重復性:通過比較相鄰稀釋梯度的代謝物檢出強度比值來評估。統計629個代謝物在兩組相鄰稀釋梯度間檢出強度比值(4×:16×、16×:64×,log2(理論值)=2),結果表明SWATHtoMRM > SWATH-MS¹> SWATH-MS²,SWATHtoMRM技術在不同的稀釋梯度組合中具有接近的中位值以及較窄的分布,同時SWATH-MS¹也是優于SWATH-MS²的,SWATH-MS²在16×:64×組合中,比值中位數已經嚴重偏離了理論值(4倍),并且具有很寬的分布范圍(圖4e)。
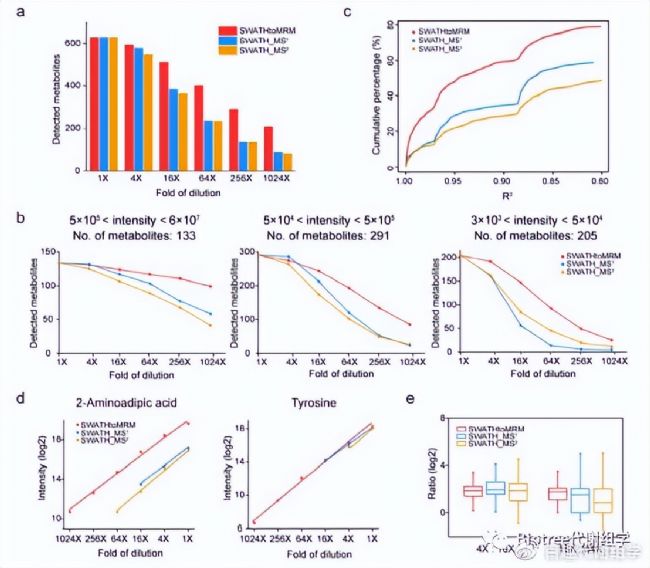
圖4. SWATHtoMRM技術定量性能評估。(a) 3種方法在不同稀釋梯度中的代謝物檢出數比較。 (b) 細化到不同豐度范圍時代謝物檢出數。(c)R²累積分布,從R²=1到R²=0.8,滿足條件的代謝物數目逐漸增多。(d) 列舉了兩種實際代謝物的定量線性范圍。(e) 兩個相鄰稀釋梯度間的比值分布圖。
SWATHtoMRM實用性評估
為了評估SWATHtoMRM技術的實用性能,作者將該技術運用于結腸癌(CRC)診斷標志物的發現研究(18對樣品作為訓練集,42對用作驗證集)。利用pooled QC樣品構建了1705個MRM離子對,并成功檢測到了其中的1303(76.4%)個代謝物。總共有1213(93.1%)個代謝物在>4個實際組織樣品中加測到,并用于后續統計分析。從QC樣品的RSD以及PCA分析結果來看,SWATHtoMRM在重復性和靈敏度方面顯著優于SWATH-MS¹技術,主要表現在更低中位RSD值和較窄的RSD分布(圖5a),以及PCA得分圖中更小的組內離散度和更大的組間分離度(圖5b)。
在生物標志物發現研究階段,1303個代謝物中有358個具有統計學差異(fold-change >1.5, p-value > 0.01),其中67個被成功鑒定。隨后,使用PLS-DA模型分析了CRC癌組織與癌旁組織的代謝物差異,VIP最高的20個代謝物被定義為潛在生物標志物(圖5d),并具有極好的辨別能力(AUC = 1)。接下來,作者在驗證集樣品中檢測了這20個代謝物,并評估了其預測精度,結果發現這些代謝物在驗證集樣品中同樣具有極好的辨別能力(AUC=0.998,95% CI)(圖5e)。
最后,作者使用CRC病人手術切除前后血漿樣品來評估了這些代謝物的預后標志物潛力。其中17個代謝物表現出可靠的預測能力(AUC = 0.779,95% CI,sensitivity = 91.2%,specificity = 64.7%)(圖5f)。
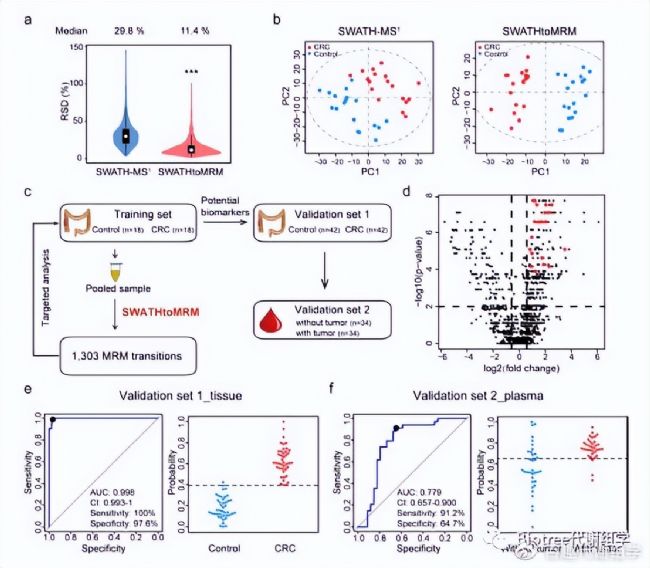
圖5. (a) QC樣品中檢測到的1213個代謝物RSD值分布。(b) 分別使用SWATH-MS¹和SWATHtoMRM技術檢測癌組織和癌旁組織中代謝物得到的PCA打分。(c)生物標志物發現研究的SWATHtoMRM實驗流程。(d) 火山圖展示1213個檢測到的代謝物,紅色點表示定義的20個潛在生物標志物。(e) 使用驗證集組織樣品的20個代謝物構建的PLS預測模型ROC曲線(左)和概率分布圖(右)。(f) 使用驗證集血漿樣品的17個代謝物構建的PLS預測模型ROC曲線(左)和概率分布圖(右)。
代謝組學文獻分享—總結
作者開發了一種新的靶向代謝組學技術—SWATHtoMRM,可同時檢測高達1000-2000個代謝物。并從多個角度詳細對比了SWATHtoMRM與DDA、SWATH-MS¹以及SWATH-MS²技術之間的優劣。與DDA相比,SWATHtoMRM技術能夠構建更多的MRM離子對。而與SWATH-MS技術相比,SWATHtoMRM具有更好的重復性、更高的靈敏度和更廣的覆蓋度。同時通過實際案例探究了該方法在代謝物生物標志物發現研究中的巨大潛力。