FB23-2šÍ·ÅūūúËØDÖúÁĶ―ŌÃØIRF8ÔÚÁÜ°Íž°û°ŨŅŠēĄÖÐĩÄŨũÓÃCÖÆ
AbMoleŌÔÆäŨŋÔ―ĩÄÆ·Ų|ĄĒÅcrūãßMĩÄŪaÆ·ĄĒĢIonĩÄ·þÕĢŽēŧāķĶÁĶÖ§ģÖČŦĮōŋÆWžŌŦ@ĩÃÖØŌŠĩÄÍŧÆÆÐÔģÉđûĄĢ
ÓÉíŨÔÉ―|īóWýRôátWÔšĩÄ Ying ZhouĢŽJingJing YeŌÔž°ChunYan JiĩČķāÃûŅÐūŋČËTÔÚÆÚŋŊAdv Sci (Weinh)ĢĻIF=15.1ĢĐÉÏĢŽđēÍŽ°ląíÁËî}é“Silencing of IRF8 Mediated by m6A Modification Promotes the Progression of T-Cell Acute Lymphoblastic Leukemia”ĩÄÎÄÕÂĄĢÔÚÔÎÄÕÂÖÐĢŽŅÐūŋČËTĘđÓÃÁËŲŨÔAbMoleĩÄFB23-2ĢĻM9422ĢĐšÍ Actinomycin DĢĻM4881ĢĐÉ·NŪaÆ·ĢŽ―ŌĘūÁËIRF8ÔÚTž°ûžąÐÔÁÜ°Íž°û°ŨŅŠēĄĢĻT-ALLĢĐÖÐĩÄŌÖÖÆŨũÓÃĢŽēĒęUÃũFTOÍĻß^Õ{ŋØIRF8ĩÄm6AžŨŧųŧŊÐÞïĢŽÄķø―é§T-ALL°lēĄĩÄūßówCÖÆĢŽéT-ALLĩÄŅÐūŋĖáđĐÁËOūßÁĶĩÄÐÂēßÂÔĄĢ

T-ALLĘĮŌŧ·NĮÖŌuÐÔŅŠŌššÐÔÄ[ÁöĢŽēĒĮŌîAšóÝ^ēîĄĢŌōīËĖ―ūŋT-ALLĩÄ°lÉú°lÕđCÖÆĢŽĪÕŌÐÂĩÄŅÐūŋ°ÐücĢŽĶđĨŋËT-ALLūßÓÐÖØŌŠĩÄŋÆWrÖĩĄĢ
ŅÐūŋČËTÍĻß^ąČÝ^T-ALLŧžēĄĘÜÔÕßŌÔž°―ĄŋĩČËÖŪégĩÄēîŪąíß_ŧųŌōĢĻDEGsĢĐĢŽ°lŽFIRF8ÔÚT-ALLŧžēĄĘÜÔÕßÖÐĘÜĩ―ŪģĢŌÖÖÆĢĻD1A-BĢĐĄĢČŧšóÍĻß^ÂýēĄķūÞDČūĢŽÔÚT-ALLž°ûÏĩMolt4šÍJurkatÖÐß^ąíß_IRF8ĢŽēĒßMÐÐCCK8šÍEdUzyĩČōĢŽ°lŽFÉÏÕ{IRF8ÄÜï@ÖøŌÖÖÆT-ALLž°ûĩÄÉúéLĢĻD1CĢĐŌÔž°ĮÖŌußwŌÆÄÜÁĶĢĻD1D-EĢĐĄĢ
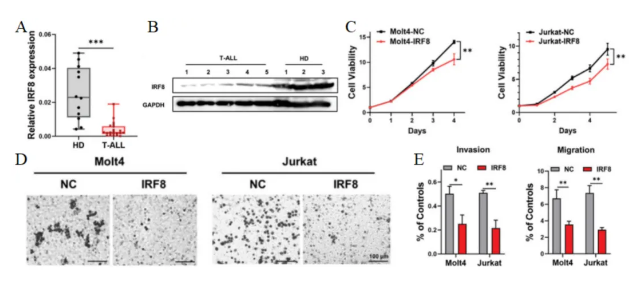
D 1 A. ÍĻß^qRT-PCRzyÐÂÔ\āĩÄT-ALLŧžēĄĘÜÔÕßšÍ―ĄŋĩđĐówđĮËčÖÐIRF8ĩÄąíß_ĄĢB. ÍĻß^ÃâŌßÓĄÛE·ĻzyÐÂÔ\āĩÄT-ALLŧžēĄĘÜÔÕßšÍ―ĄŋĩđĐówđĮËčÖÐIRF8ĩÄąíß_ĄĢC. ÍĻß^CCK8Ôō·ÖÎöIRF8ąíß_ĩÄMolt4ž°ûĄĒJurkatž°ûšÍNCsĩÄÔöÖģĮérĄĢD. ß^ąíß_IRF8ĩÄMolt4šÍJurkatž°ûĢŽŌÔž°NCž°ûÔÚMatrigelŧųŲ|ÄĪÏÂĩÄ―Yū§ŨÏČūÉŦĄĢE. ß^ąíß_IRF8ĩÄMolt4ž°ûšÍNCĩÄĮÖŌušÍßwŌÆÄÜÁĶĄĢ
―ÓÏÂíĢŽŅÐūŋČËTÍĻß^―ĻIrf8ĮÃģýÐĄĘóßMÐÐōĢĻD2AĢĐĢŽ―Yđûï@ĘūĢŽIrf8-/-―MÐĄĘóĩÄT-ALL°lÕđūßÓÐļßĮÖŌuÐÔĢŽēĒĮŌ·üÆÚļüķĖĢŽÍŽrąíŽFģöÃũï@ĩÄļÎÆĒÄ[īóšÍđÉđĮÉn°ŨĢŽÕfÃũīæÔÚ°Ũž°ûÔöķāĄĒžtž°ûpÉŲšÍķāÆũđŲ―þĩČĮérĢĻD2BĢĐĄĢīËÍâĢŽŧųŌōĮÃģýÐĄĘóđĮËčGFP+ž°ûÖÐKi67ËŪÆ―ÉýļßĢŽąíÃũĮÃģýIrf8ÄÜīŲßM°ŨŅŠēĄž°ûĩÄÔöÖģĢĻD2CĢĐĢŽĖáĘūIrf8ĩÄȹʧÄÜÅcōÓŧųŌō fÍŽŨũÓÃĢŽđēÍŽžÓËŲT-ALL°lÕđĄĢ
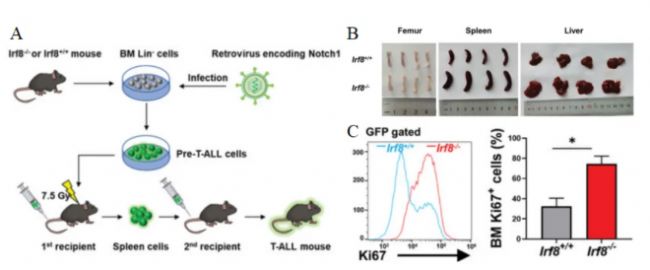
D 2 A. ―ĻÁĒT-ALLÐĄĘóÄĢÐÍĩÄģĖÐōĄĢB. ŌÆÖēÁËIrf8-/-šÍIrf8+/+ÆĒž°ûĩÄÐĄĘóÔÚŌÆÖēšóÉÖÜrĩÄđÉđĮĄĒÆĒÅKšÍļÎÅKDÏņĄĢC. ŌÆÖēÉÖÜšóĢŽIrf8-/-―MšÍIrf8+/+―MGFP+éTŋØđĮËčž°ûĩÄKi67ąíß_ĮérĄĢ
KEGGĶDEGsĩÄ·ÖÎöï@ĘūĢŽPI3K/AKTÐÅĖÍĻ·ï@ÖøļŧžŊĢĻD3AĢĐĄĢëSšóßxņPI3K/AKTÐÅĖÏāęPDEGs—PIK3R5ĢĻD3BĢĐßMÐÐßMŌŧē―ŅÐūŋĄĢÃâŌßÓĄÛEĢĻD3CĢĐšÍCCK8ĢĻD3DĢĐ―Yđûï@ĘūĢŽÔÚIRF8ß^ąíß_ĩÄMolt4ž°ûÖÐĢŽPIK3R5ąíß_ÏÂÕ{ĢŽAKTšÍmTORĩÄÁŨËáŧŊËŪÆ――ĩĩÍĄĢąíÃũÔÚT-ALLÖÐĢŽIRF8ÍĻß^PIK3R5ØÕ{ŋØPI3K/AKTÍĻ·ĄĢ
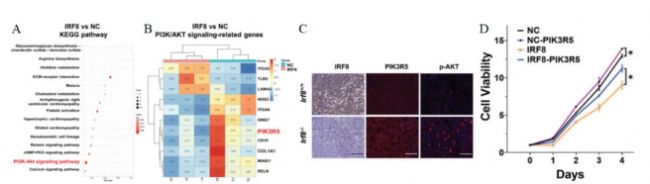
D 3 A. KEGG·ÖÎöï@ĘūĢŽÅcęÐÔĶÕÕĢĻNCĢĐMolt4ž°ûÏāąČĢŽRNA-seqzyĩ―IRF8ąíß_ĩÄMolt4ž°ûÖÐPI3K/AKTÍĻ·ļŧžŊĄĢB. áDï@ĘūÁËIRF8ąíß_ĩÄMolt4ž°ûĢĻE1ĄĒF1ĄĒG1ĢĐšÍęÐÔĶÕÕĢĻNCĢĐMolt4ž°ûĢĻE2ĄĒF2ĄĒG2ĢĐÖÐÅcPI3K/AKTÐÅĖÏāęPĩÄDEGsĄĢPIK3R5ąŧī_ķĻéDEGĄĢC. Irf8-/-šÍIrf8+/+ T-ALLÐĄĘóÆĒÅKÖÐIRF8ĄĒPIK3R5šÍp-AKTąíß_ĩÄÃâŌßÉđâ―YđûĄĢD. PIK3R5ß^ąíß_ĶMolt4-NCšÍMolt4-IRF8ž°ûŧîÁĶĩÄÓ°íĄĢ
ÖŪšóĢŽŅÐūŋČËTĘđÓÃJasparĩþėĢŽÔÚPIK3R5ĩÄĒÓŨÓ ^Óō°lŽFIRF8ĩÄÔÚ―YšÏÎŧücĢĻD4AĢĐĄĢČūÉŦŲ|ÃâŌßģÁĩíĢĻChIPĢĐï@ĘūĢŽIRF8ÃâŌßģÁĩíÖÐĩÄPIK3R5ĒÓŨÓÐōÁÐï@ÖøļŧžŊĢĻD4BĢĐĢŽąíÃũIRF8ŋÉÖą―ÓŨRePIK3R5ĒÓŨÓÐōÁÐÖÐĩÄ―YšÏŧųÐōĢŽÕ{ŋØPIK3R5ÞDäĄĢ

D 4 A. JasparĩþėîAyĩÄPIK3R5ĒÓŨÓ ^ÓōÖÐŋÉÅcIRF8―YšÏĩÄÔÚŧųFĄĢB. Molt4ž°ûÖÐPIK3R5ĩÄChIPĐ\qPCR·ÖÎöšÍÏāŠĩÄëÓūDŨVĄĢ
ëSšóĢŽŅÐūŋČËTĶT-ALLĩþžŊGSE13159ßMÐÐŧųŌō―MļŧžŊ·ÖÎöĢĻGSEAĢĐĢŽ―Yđûï@ĘūT-ALLŧžēĄĘÜÔÕßówČļũ·Nm6AÕ{ŋØŌōŨÓ°lÉúï@ÖøŨŧŊĢŽÆäÖÐŌÔFTOĩÄŨŧŊËŪÆ―ŨîéÃũï@ĢĻD5AĢĐĄĢPearsonÏāęP·ÖÎöąíÃũĢŽÔÚÉŠÁĒĩÄT-ALLČšówÖÐĢŽFTOšÍIRF8ĩÄąíß_īæÔÚÃũï@ĩÄØÏāęPÐÔĢĻD5BĢĐĄĢ
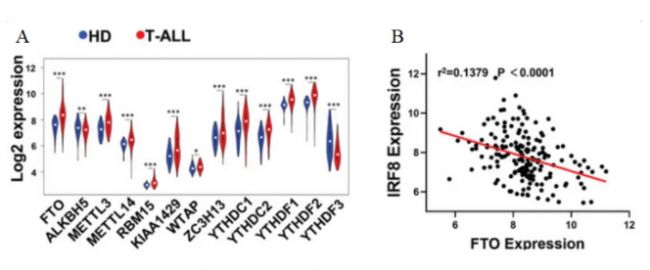
D 5 A. T-ALLŧžēĄĘÜÔÕßšÍ―ĄŋĩđĐówĢĻHDsĢĐđĮËčÖÐm6AÕ{đŌōŨÓĩÄLog2ąíß_ÖĩĄĢB. íŨÔGSE13159ĩÄ174·ÝT-ALLÓąūÖÐFTOšÍIRF8ąíß_ËŪÆ―ĩÄÏāęPÐÔĢŽPearsonÏāęPÐÔ·ÖÎöĄĢ
―ÓÏÂíĢŽÓÃÐĄ°lARNAĢĻshRNAĢĐÂýēĄķūĮÃģýFTOŧōĘđÓÃFB23-2ŌÖÖÆFTOËŪÆ―ßMÐÐōĢŽzy°lŽFĢŽÉ·N·―Ę―ūųþ§ÖÂIRF8ąíß_ËŪÆ―ÉýļßĢĻD6A-BĢĐĢŽŨCÃũIRF8ĩÄąíß_ĘÜĩ―FTO―é§ĩÄm6AÐÞïĩÄØÕ{ŋØĄĢ
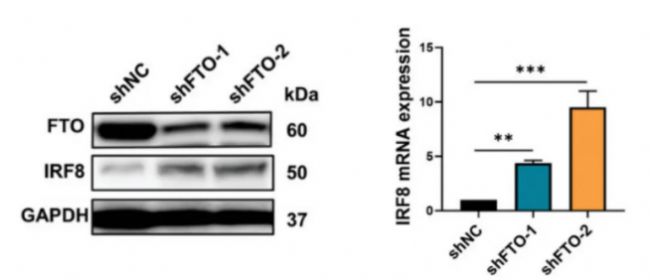
D 6 A. ÍĻß^WesternÓĄÛEzyÞDČūFTO shRNAÂýēĄķūĢĻshFTO-1šÍshFTO-2ĢĐŧōęÐÔĶÕÕĢĻshNCĢĐĩÄMolt4ž°ûÖÐFTOšÍIRF8ĩÄËŪÆ―ĄĢB. ÞDČūshFTO-1ĄĒshFTO-2ŧōshNCÂýēĄķūĩÄMolt4ž°ûÖÐIRF8 mRNAËŪÆ―ĩÄqRT-PCR·ÖÎöĄĢ
ëSšóĢŽŅÐūŋČËTĘđÓÃFB23-2ĖĀíMolt4ž°ûĢŽ°lŽFm6AĩÄŋówØSķČĖáļßĢĻD7AĢĐĢŽĮŌIRF8ĩÄąíß_Ėáļß2ÖÁ3ąķĢĻD7BĢĐĄĢMeRIP-seq·ÖÎöï@ĘūĢŽFTOʧŧî§ÖÂIRF8 3′·Į·Ũg ^ĢĻUTRĢĐĩÄm6AËŪÆ―Ãũï@ÔöžÓĄĢīËÍâĢŽFTO RIP-seq·ÖÎöï@ĘūĢŽFTO―YšÏ·åļŧžŊÔÚIRF8 mRNAÞDäąūÖÐĢŽIRF8ÞDäąū3′UTRÖÐĩÄÜÛEÅcm6A·åÖØŊBĢĻD7CĢĐĄĢ
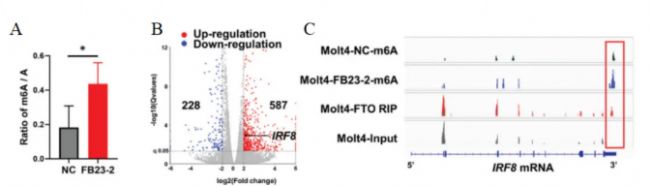
D 7 A. ÓÃ2 µm FB23-2ŧōDMSOĢĻNCĢĐÅāðB48ÐĄrĩÄMolt4ž°ûÖÐmRNAĩÄŋówm6AØSķČĢŽēĒÓÃLC-MS/MSÃčĘöm6A/AĩÄąČÂĘĄĢB. DEGsĩÄŧðÉ―DĢŧIRF8ÓÚÚücËģöĄĢË{ücĢšÏÂÕ{ŧųŌōĢŧžtücĢšÉÏÕ{ŧųŌōĄĢC. Integrative Genomics Viewer (IGV)Ũ·ÛÃčĀLÓÃDMSOÝdówŧōFB23-2ĖĀíĩÄMolt4ž°ûÖÐIRF8ŧųŌōĩÄm6AÐÞïĢŽŌÔž°IRF8ŧųŌōŨųÖÐĩÄFTO RIP-seqÐÅĖšÍÏāŠĩÄÝČëÐÅĖĄĢ
ÖŪšóÔOÓŌýÎïUÔöIRF8 3′UTRÖаüšŽîAyĩÄÔÚ―YšÏÎŧücĩÄÐōÁÐĢĻD8AĢĐĢŽēĒßMÐÐRIP-qPCRzyĢŽđēšYßxģöÎåŋÉÄÜąŧFTOÖą―Ó―YšÏĩÄÔÚm6AÎŧücĢĻD8BĢĐĄĢëSšóĢŽĒšōßxm6AÎŧücÄAĩ―T·ÖeĖæQĢĻMUT1ÖÁMUT5ĢŽD8CĢĐĢŽēĒ―ĻŌ°ÉúÐÍšÍÎåÍŧŨÐÍIRF8 3′UTRóļæÝdówĄĢ―ŅÐūŋ°lŽFĢŽÞDČūWTóļæÝdówž°ûĩÄÉđâËØÃļŧîÐÔÃũï@ļßÓÚHEK293T-shNCž°ûĄĢēĒĮŌÞDČūMUT1ÖÁMUT5óļæÝdówšóĢŽŋÉŌÔŧÖÍÉđâËØÃļŧîÐÔĢĻD8DĢĐĄĢąíÃũIRF8ĘĮFTOĩÄŌŧęPæI°ÐücĢŽFTOÍĻß^ß@ÐĐm6AÎŧücØÕ{ŋØIRF8 mRNAĩÄąíß_ĄĢëSšóĢŽŅÐūŋČËTĘđÓÃFB23-2šÍActinomycin DĖĀíMolt4šÍJurkatž°ûĢŽ°lŽFIRF8ÞDäąūĩÄ°ëËĨÆÚÃũï@ŅÓéLĢĻD8EĢĐĄĢąíÃũŌÖÖÆFTOĩÄČĨžŨŧųŧŊŧîÐÔŋÉÍĻß^īŲßMRNA·ķĻÐÔšÍpÉŲRNA―ĩ―âíŧÖÍIRF8 mRNAĩÄąíß_ĄĢ
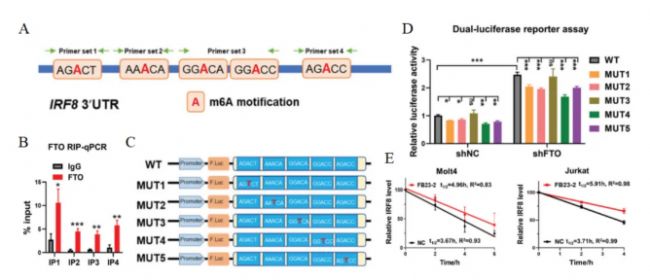
D 8 A. IRF8 3′ UTRÖÐÔÚĩÄm6A―YšÏÎŧücĢĻÎŧüc1-5ĢĐĘūŌâDĄĢŌýÎï―M3ĢĻIP3ĢĐ°üĀĻÉÎŧücĢšÎŧüc3šÍÎŧüc4ĄĢB. FTO RIP-qPCR·ÖÎöï@ĘūFTOÅcMolt4ž°ûÖÐIRF8 mRNAÞDäąū―YšÏĄĢC. ŋËÂĄĩ―pMIR-REPORTÉđâËØÃļÝdówĩÄIRF8 mRNA 3′ UTRŌ°ÉúÐÍĢĻWTĢĐŌÔž°m6AŨReÎŧücÄAšËÜÕËáÍŧŨéTšËÜÕËáĩÄÎåÍŧŨÐōÁÐĢĻMUT1ÖÁMUT5ĢĐĘūŌâDĄĢD. Ō°ÉúÐÍIRF8 mRNA 3′ UTRĢĻWTĢĐšÍÍŧŨÐÍIRF8 mRNA 3′ UTRĢĻMUT1ÖÁMUT5ĢĐóļæÝdówÞDČūÓÐŧōoFTOĮÃģýĩÄHEK293Tž°ûĩÄÏāĶÉđâËØÃļŧîÐÔĄĢE. ÓÃ5 µm FB23-2ŧōDMSOĢĻNCĢĐîAĖĀíMolt4šÍJurkatž°û24ÐĄrĢŽēĒÔÚÖļķĻrégÓÃ5 µg/mL Actinomycin DÅāðBĩÄIRF8 mRNA―ĩ―âzyĢŽēĒÅc0ÐĄrĩÄmRNAËŪÆ―wŌŧŧŊĄĢ
ŨîšóŅÐūŋČËTĘđÓÃFB23-2ĖĀíIrf8+/+―MšÍIrf8-/-―MĩÄT-ALLÐĄĘóÄĢÐÍĢĻD9ĢĐĢŽēĒÍĻß^ßMÐÐÁũĘ―ž°ûÐgĢŽücÓĄÛEzyŌÔž°ÃâŌßÓĄÛEĩČ·ÖÎöĄĢ―YđûąíÃũĢŽŌÖÖÆFTOĩÄm6AČĨžŨŧųŧŊÃļŧîÐÔĢŽÄÜŧÖÍT-ALLÖÐIRF8ĩÄąíß_ĢŽÄķøÍĻß^PI3K/AKTÐÅĖÞD§ŌÖÖÆT-ALLĩÄ°lÉúĄĢ

D 9 ―ĻÁĒIrf8-/-šÍIrf8+/+ T-ALLÐĄĘóÄĢÐÍž°FB23-2―oËrégąíĢĻFB23-2Ģš2 mg/kgĢĐĄĢ
ÔŅÐūŋĘđÓÃÁËŲŨÔAbMoleĩÄFB23-2ĢĻM9422ĢĐšÍ ActinomycinDĢĻM4881ĢĐÉ·NŪaÆ·ĢŽFB23-2ĘĮŌŧ·NÐÂÐÍFTOÐĄ·ÖŨÓŌÖÖÆĐĢŽŋÉÖą―ÓÅcFTO―YšÏĢŽßxņÐÔĩØŌÖÖÆm6AČĨžŨŧųŧŊÃļĩÄŧîÐÔĢŽÔÚÍâï@ŨÓRNAžŨŧųŧŊ°ÐÏōŅÐūŋîIÓōūßÓÐūÞīóĩÄÁĶšÍÝĄĢÔÚÔōÖÐĢŽŅÐūŋČËTķāīÎĘđÓÃFB23-2ŌÖÖÆFTOŧîÐÔĢŽßMķøļüšÃĩÄŅÐūŋFTOÅcIRF8ąíß_ÖŪégĩÄÕ{ŋØCÖÆĄĢActinomycin DĘĮŌŧ·NÞDäŌÖÖÆĐĢŽÔÚÔōÖÐÓÃÓÚÔÚ mRNA―ĩ―â·ÖÎöōÖÐŌÖÖÆÞDäĢŽąÜÃâÐÂÞDäĩÄmRNAÓ°íō―YđûĄĢ
ŅÐūŋČËTĘđÓÃFB23-2ŌÖÖÆFTOĩÄŧîÐÔĢŽ°lŽFIRF8ĩÄąíß_ËŪÆ―ÉýļßĢŽž°ûÔöÖģĘÜĩ―FB23-2ĐÁŋŌĀŲÐÔŌÖÖÆĢĻD10A-BĢĐĢŽēĒŋÉŌÔÍĻß^ĮÃģýIRF8ĩÃĩ―ÓÐЧū―âĄĢßMŌŧē―ŅÐūŋ°lŽFĢŽFB23-2ĖĀíšÍIRF8ĮÃģýÖŪégÔÚž°ûŧîÁĶĢĻD10CĢК͞ŊÂäÐÎģÉŧîÐÔĢĻD10DĢĐ·―ÃæūßÓÐÃũï@ĩÄ―ŧŧĨŨũÓÃĢŽÕfÃũIRF8ĩÄąíß_ĘÜĩ―FTO―é§ĩÄm6AÐÞïĩÄØÕ{ŋØĄĢ
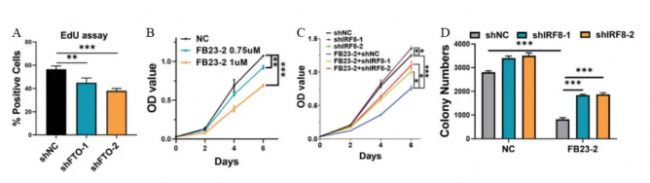
D 10 A. ÓÃēŧÍŽâķČĩÄFB23-2ŧōDMSOĢĻNCĢĐĖĀíMolt4ž°û48ÐĄrĩÄIRF8 mRNAËŪÆ―ĄĢB. ÓÃēŧÍŽâķČĩÄFB23-2ŧōDMSOĢĻNCĢĐĖĀíĩÄMolt4ž°ûĩÄÔöÖģĮérĄĢC. ÓÃshIRF8-1ĄĒshIRF8-2ŧōshNCÂýēĄķūÞDČūĩÄMolt4ž°û―FB23-2ĖĀíšóĩÄŧîÁĶĄĢD. ÞDČūshIRF8-1ĄĒshIRF8-2ŧōshNCÂýēĄķūēĒ―1 µm FB23-2ĖĀíĩÄMolt4ž°ûĩÄžŊÂäÐÎģÉÄÜÁĶĄĢ
īËÍâĢŽŅÐūŋČËTÍĻß^ĶIrf8+/+―MšÍIrf8-/-―MĩÄT-ALLÐĄĘóßMÐÐFB23-2ĖĀíĢĻD9ĢĐĄĢÁũĘ―ž°ûÐgï@ĘūĢŽ―oËFB23-2ÄÜï@Öø―ĩĩÍIrf8+/+―MđĮËčĄĒŅŠŌššÍÆĒÅKÖÐGFP+ž°ûĩÄąČĀýĢŽŌÔž°Ki67ĩÄËŪÆ―ĢĻD11AĢĐĢŽēĒĮŌÐĄĘóĩÄīæŧîrégŅÓéLĢĻD11BĢĐĄĢÕfÃũFB23-2ÄÜÓÐЧpÝpIrf8+/+T-ALLÐĄĘóĩÄ°ŨŅŠēĄØúēĒŅÓéLÆäÉúīæÆÚĄĢÃâŌßÓĄÛEšÍÃâŌßÉđâ·ÖÎöąíÃũĢŽFB23-2ÄÜï@ÖøÔöžÓIrf8+/+―MÐĄĘóđĮËčšÍÆĒÅK°ŨŅŠēĄž°ûÖÐIRF8ĩÄąíß_ĢŽēĒŌÖÖÆPIK3R5ĩÄąíß_šÍAKTĩÄÁŨËáŧŊĢĻD11C-DĢĐĄĢŌōīËĢŽŅÐūŋČËTÕJéFB23-2ŌÖÖÆFTOĩÄm6AČĨžŨŧųŧŊÃļŧîÐÔĢŽÄÜģÉđĶŧÖÍT-ALLÖÐIRF8ĩÄąíß_ĢŽēĒÄÜÍĻß^ŌÖÖÆPI3K/AKTÐÅĖÞD§ĢŽÄķøŌÖÖÆ°ŨŅŠēĄĩÄ°lÉúß^ģĖĄĢ
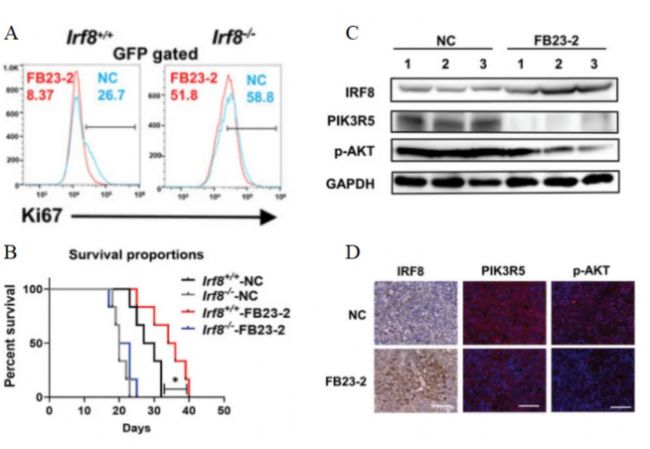
D 11 A. ÁũĘ―ž°ûxyķĻĩÄGFP+éTŋØđĮËčž°ûĩÄKi67ąíß_ËŪÆ―ĄĢB. ÓÃFB23-2ŧōDMSOĢĻNCĢĐĖĀíĩÄIrf8-/-―MšÍIrf8+/+―MÐĄĘóĩÄīæŧîÂĘĄĢC. ÃâŌßÓĄÛE·ÖÎöFB23-2ŧōDMSOĢĻNCĢĐĖĀíĩÄIrf8+/+T-ALLÐĄĘóđĮËčÖÐĩÄIRF8ĄĒPIK3R5šÍp-AKTËŪÆ―ĄĢD. ―oËFB23-2ŧōDMSOĢĻNCĢĐšóIrf8+/+T-ALLÐĄĘóÆĒÅKÖÐIRF8ĄĒPIK3R5šÍp-AKTąíß_ĩÄÃâŌßÉđâDÆŽĄĢ
ŋÖŪĢŽÔŅÐūŋ―ŌĘūÁËŌŧ·NÐÂĩÄT-ALLŧųŌōÕ{ŋØCÖÆĢŽēĒéT-ALLĩÄßzũCÖƚͰÐÏōŅÐūŋĖáđĐÁËÐÂĩÄŌ―ĮĄĢ
ūīÕęPŨĒĢšwww.abmole.cn

- GelNEST™ŧųŲ|ÄzÖúÁĶŠĶŲQŌŨąÚūÏÂĩÄŋÆŅÐēŧī_ķĻÐÔ
- ĀÂüžžÐgÔÚÎīíátWÖÐĩÄŠÓÃátđĪČÚšÏÉģýģÉđĶÅeÐÐ
- ēĐúōvÔĐ2025ĘŨīÎīóīŲäNé_ĒĢŽģŽķāŧÝĩČÄãí
- ÖÐĖÐÂÖÛé_WŧÝ:ž°ûōĖŨŅb980ÔŠ,ÅāðBŧųÕÛŋÛĩČ
- ÖÐĖÐÂÖÛÏÞrīŲäNĢšŲIž°ûūÍËÍÖ§ÔówzyÔĐšÐ
- ČAÍþÖÐxÔĐšÐÄęÄĐÏÞrŧÝíŌuĢŽĶÖĩÏíšÃķY
- ÃĀĩÂÂÍÆģöÐÂÆ·ÖØ―Mž°ûÏĩÓÃÓÚÉúŪaÎŋËÂĄŅŠÐÍÔĐ
- °ŲÎÐĄûÅßŅŋÄýžŊËØĖ―áČūÁÏ8ÕÛŧÝĢŽŲIžīŲĘóË|