五條腫瘤細胞代謝通路的機制原理及與腫瘤微環境的關系研究
以下文章來源于閑談 Immunology ,作者谷語
即使在氧充分的條件下(有氧糖酵解/Warburg效應),癌細胞上調葡萄糖的糖酵解途徑形成乳酸,這個過程被認為是惡性腫瘤的標志。代謝活躍的癌細胞導致營養消耗、缺氧、酸性、有毒的代謝物富集的腫瘤微環境,抑制免疫系統。
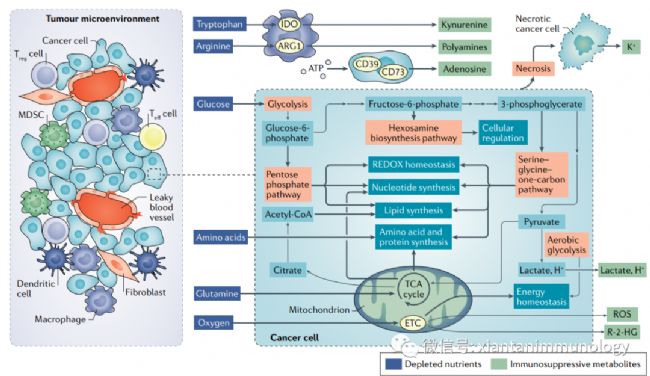
腫瘤細胞代謝與腫瘤微環境(Nat Rev Cancer . 2020 Sep;20(9):516-531)
1、Hippo Pathway Hippo通路控制細胞增殖、器官大小和組織穩態。它由MAPK家族、MST1/2、LATS1/2以及轉錄共激活因子YAP和TAZ組成。 當該通路被激活時,上游信號磷酸化并激活MST1/2和LATS1/2,從而磷酸化YAP/TAZ。因此,YAP/TAZ通過14-3-3蛋白被隔離在細胞質中,并被E3連接酶β-TRCP降解。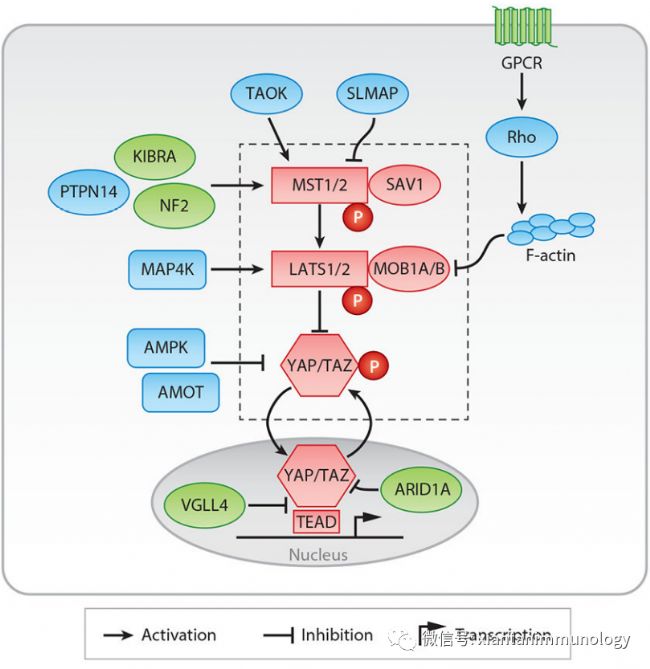
Annu Rev Pathol. 2021 January 24; 16: 299–322
然而,當Hippo信號通路關閉時,YAP/TAZ易位到細胞核,與轉錄增強相關域(TEAD)轉錄因子結合,誘導致癌靶基因的表達。
Hippo通路受到廣泛的上游調控因子的調控,包括細胞-細胞接觸、來自周圍環境刺激、Wnt信號傳導、GPCR-配體相互作用和各種細胞應激條件。
YAP/TAZ促進糖酵解和谷氨酸代謝
YAP/TAZ活性在許多類型的癌癥中都是過度活躍。hHippo通路作為細胞生長的主調節因子,參與了多種代謝過程。YAP/TAZ活性通過直接和間接提高糖酵解酶活性來促進糖酵解。特別是,YAP/TAZ的活性增加了TEAD對GLUT3的表達,并通過FOXC2誘導了HK2的表達。YAP/TAZ還通過Hedgehog信號通路促進LncRNABCAR4的表達,上調HK2和PFKFB3。YAP/TAZ增強谷氨酰胺代謝,增加乳腺癌細胞中谷氨酰胺轉運體SLC1A5和SLC7A5的表達。YAP/TAZ和TEAD通過表達氨基酸轉運體上調谷氨酰胺和氨基酸代謝。YAP/TAZ誘導谷氨酰胺酶和轉氨酶的表達,包括GOT1和PSAT1,它們產生NEAAs和TCA循環中間體。YAP/TAZ積累脂質,直接調節膽汁酸成分,從而增強癌細胞的轉移潛能。
葡萄糖增強YAP/TAZ的活性
細胞周期進程和細胞增殖由營養物質濃度決定。葡萄糖代謝對YAP/TAZ活性有顯著影響。高糖水平增加了HSP的葡萄糖通量,產生用于糖基化的UDP-GlcNAc。因此,豐富的葡萄糖誘導YAP O-GlcNAcylation,干擾LATS和βTrCP的相互作用,進而增強YAP/TAZ的活性。在胰腺癌和肝癌中,YAP被O-GlcNAcylation過度激活。
LATS2的糖基化通過干擾乳腺癌中LATS2和MOB1的相互作用來抑制其活性。相反,葡萄糖剝奪誘導的能量應激通過hippo依賴和非依賴的機制抑制YAP/TAZ活性。能量傳感器AMPK隨著ATP的減少而被激活。AMPK直接磷酸化YAP的絲氨酸61和絲氨酸94,從而阻斷YAP-TEAD的相互作用。AMPK通過AMOTL1磷酸化和激活間接抑制YAP。外部激素水平可以調節Hippo通路。脂質激素,如溶血磷脂酸和鞘氨醇-1-磷酸,通過GPCRs抑制該通路,肽激素胰高血糖素增加血糖水平,通過cAMP和PKA激活Hippo通路。
脂肪增強YAP/TAZ的活性
亞細胞脂質成分也可以調節Hippo通路激酶。SREBP是Hippo通路的上游調控因子。當SREBP被激活時,甲羥戊酸途徑增強了RhoAGTPase的香葉酰化。RhoA是一個f-actin的細胞骨架調節因子,而f-actin是一個成熟的LATS激酶的上游因子。因此,癌細胞中脂肪酸代謝的增加會異常激活RhoA,并通過抑制LATS來增強YAP/TAZ的活性。有趣的是,YAP/TAZ在血液系統惡性腫瘤中幾乎不表達,而YAP的強制表達已被證明可以介導腫瘤抑制功能。
在正常細胞中,細胞的增殖和合成代謝受到外部生長因子(如胰島素和激素)的微妙調節,PI3K-AKT-mTOR通路是其中一條重要通路。 PI3K/AKT信號通路調節代謝酶。在被胰島素激活后,AKT通過磷酸化GSK3來抑制糖原合成途徑。PI3K/AKT通過代謝酶的翻譯后修飾(如磷酸化、糖基化)直接或間接上調糖酵解。例如,AKT磷酸化并激活AS160蛋白,從而增強了GLUT的膜運輸。AKT通過抑制磷酸化直接抑制TXNIP,通過抑制內吞作用增加GLUT1/4的膜表達。 AKT還提高了糖酵解酶的效率。AKT通過增加HK2的線粒體整合來磷酸化并激活HK2。AKT使PFKFB磷酸化;這種變化催化果糖-6-磷酸到F-2,6-BP,從而提高糖酵解。AKT信號通路也通過HIF-1α轉錄因子促進糖酵解。HIF-1激活一些糖酵解組分,包括GLUT。HIF-1誘導LDH促進乳酸分泌和有氧糖酵解。 PI3K/AKT信號通路上調谷氨酰胺和脂肪酸代謝。AKT通過mTORC1增加Myc的翻譯,并通過GSK3和FOXO3A抑制Myc的降解。由于MYC調節谷氨酰胺代謝,MYC的激活增加了細胞氮的供應。因此,AKT促進了核苷酸的合成。癌細胞需要從頭脂質合成來維持和供應持續的無限細胞分裂所需的膜成分。AKT通過直接磷酸化脂質合成的第一個酶ACLY,影響脂質的新生生物合成過程,從而提高酶的效率。AKT通過ACLY調控增加各種癌癥類型中的組蛋白乙酰化 AKT誘導的mTORC1激活增強了SREBP家族的翻譯和切割加工。AKT介導的GSK3抑制進一步阻止了SREBP1的降解。因此,了解通過PI3K/AKT信號通路的代謝重編程將闡明PI3K抑制劑對癌癥治療的有效治療替代方案。
3、Myc Pathway轉錄因子c-Myc通過MAX調控基因表達,是癌細胞中最過度活躍的基因之一,位于生長相關信號通路(如EGFR、AKT和GPCRs)的交叉點。
Myc調節與葡萄糖、谷氨酰胺和脂肪酸代謝相關的基因的表達。Myc通過上調糖酵解和谷氨酰胺水解來增強癌細胞的代謝重。Myc誘導許多糖酵解酶(如GLUT、HK2和PFK),它們催化糖酵解過程相應步驟。
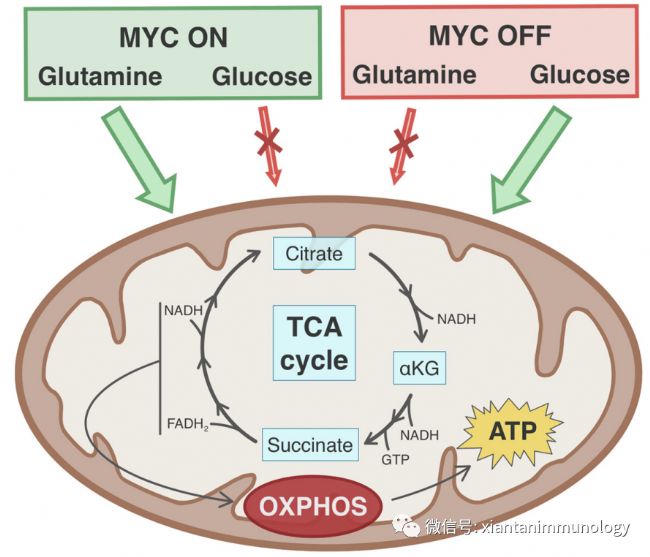
MYC癌基因的激活驅動谷氨酰胺的優先代謝向TCA循環添補反應(Anaplerosis)和隨后的氧化磷酸化。MYC癌基因表達的缺失增加了線粒體代謝對葡萄糖的依賴。
P53是最著名的腫瘤抑制基因之一,它影響細胞過程,包括凋亡、細胞周期進程和代謝。p53在細胞應激條件下被激活,如DNA損傷和營養剝奪。根據應激源的類型和強度,它決定了是發生適應還是細胞死亡。p53在轉錄、翻譯和翻譯后水平上被調節。p53的穩定性受E3連接酶MDM2的調控。
AMPK磷酸化,乙酰化,從而在能量應激條件下穩定p53。通過上調MDM2來促進p53的降解,p53誘導一個負反饋回路來維持穩態。
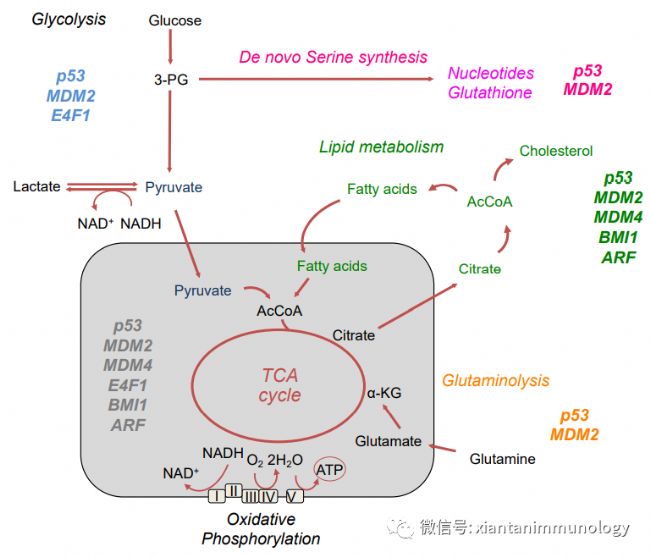
p53調節代謝(Cancers 2021, 13, 133)P53減少有氧糖酵解,上調線粒體分解代謝過程,包括脂肪酸氧化(FAO)和OXPHOS;p53轉錄抑制GLUT1和GLUT4的表達,抑制葡萄糖攝取;抑制TCA循環;增加脂肪酸氧化;增加氧化磷酸化(OXPHOS),積累NADH和FADH2。
5. LKB1/AMPK Pathway
AMPK是一種保守性良好的能量傳感激酶。AMPK抑制合成代謝途徑(脂肪酸合成、mTOR途徑),促進分解代謝(糖酵解、Krebs循環、磷酸戊糖途徑、脂肪酸氧化和自噬)的細胞過程。因此,AMPK的激活允許細胞承受細胞能量應激條件。
與其他代謝蛋白不同,AMPK的調控依賴于AMP和ADP的細胞濃度,但獨立于其他代謝中間體。AMP/ATP比值的增加導致AMP相互作用,隨后是AMPK的構象變化。在癌癥的背景下,應激環境下的AMPK激活,包括缺氧和能量或營養剝奪,賦予了各種癌癥的應激抵抗特性。

*紅色箭頭(激活上調)(Oxid Med Cell Longev . 2019 Oct 31;2019:8730816.)
AMPK是一種致癌蛋白還是腫瘤抑制蛋白仍有待確定。在臨床環境中,AMPK激活劑,包括2型糖尿病藥物二甲雙胍和AICAR,被用于抑制癌細胞增殖;然而,AMPK也通過促進原蛋白抗性、遷移和轉移作為致癌蛋白。 ACC是AMPK的一個成熟的靶點。ACC在脂肪酸從頭合成過程中,ACC介導乙酰輔酶A轉化為丙二酰輔酶A。AMPK通過抑制ACC磷酸化來減少ATP的消耗。AMPK降低的脂肪酸合成通量降低了NADPH的消耗。為了滿足細胞對脂質代謝物的需求,AMPK通過CD36和其他脂質轉運體上調脂肪酸的攝取來維持脂質穩態。
蛋白質翻譯是高度耗能的,并使用多種合成代謝途徑,包括PPP和谷氨酰胺水解。AMPK通過抑制mTORC1和翻譯機制,允許癌細胞適應惡劣的營養條件,如葡萄糖剝奪。
主要參考文獻
- Jordan H. Driskill et al,The Hippo Pathway in Liver Homeostasis and Pathophysiology. Annu Rev Pathol. 2021 January 24; 16: 299–322
- Robert D. Leone and Jonathan D. Powell, Metabolism of immune cells in cancer, Nat Rev Cancer . 2020 Sep;20(9):516-531
- Tambay, V.; Raymond, V.-A.; Bilodeau, M. MYC Rules: Leading Glutamine Metabolism toward a Distinct Cancer Cell Phenotype. Cancers 2021, 13, 4484: Lahalle, A.; Lacroix, M.; De Blasio, C.; Cissé, M.Y.; Linares, L.K.; Le Cam, L. The p53 Pathway and Metabolism: The Tree That Hides the Forest. Cancers 2021, 13, 133
- Francesco Ciccarese et al. LKB1/AMPK Pathway and Drug Response in Cancer: A Therapeutic Perspective,Oxid Med Cell Longev . 2019 Oct 31;2019:8730816.