尚未成藥靶點的開發新方法及在治療疾病中的作用

 Tips:
Tips:
在過去的幾十年里,人們發現有些生物大分子如激酶、受體和離子通道等,與疾病發展密切相關,且具有與配體結合的特異性疏水口袋以及結合后的功能變化。便把這類靶點稱為“可成藥靶點”。
另一方面,越來越多與疾病相關的靶點被發現缺乏傳統可藥性靶點的特征,如靶蛋白的功能界面平坦,缺乏配體相互作用口袋,使得藥物設計面臨巨大挑戰,便稱為“不可成藥”靶點。例如一些典型的不可藥性靶點包括小 GTP 酶 (如 KRAS)、磷酸酶、轉錄因子、表觀遺傳靶點和蛋白質-蛋白質相互作用 (PPIs)等[1]。然而這些“不可藥性”靶點在治療人類疾病中發揮重要作用,逐漸引起醫學領域重視。尤其在 2021 年, KRASG12C 抑制劑 Sotorasib 獲得了 FDA 批準用于非小細胞肺癌患者的治療,打破了人們對不可成藥靶點的認識,即這些“不可成藥靶點”其實是“難以成藥”或“尚未成藥”靶點,通過一定的方法也可以開發出對應的藥物。

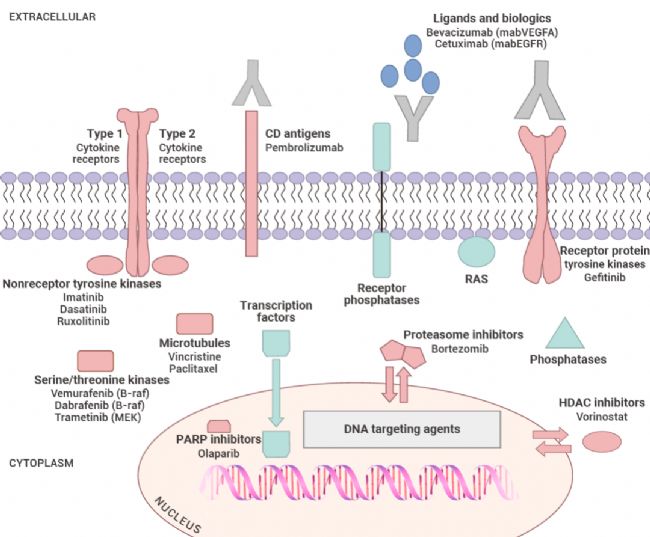
圖 1. 當代癌癥藥物分子靶點的示意圖[2]。
目前針對“不可成藥靶點”常用的藥物開發方法包括雙功能分子、共價抑制劑、變構抑制劑、PPI 藥物、基因治療等,并取得了一定的臨床成果。

雙功能分子一般是利用泛素-蛋白酶體系統或溶酶體系統實現對靶蛋白的降解。其中最近火出圈的 PROTAC 分子就是利用泛素-蛋白酶體系統實現對胞內蛋白的特異性降解,可以靶向不可成藥靶點,且具有解決耐藥性問題等優勢,在癌癥及其他疾病治療中表現出強大的潛力。(詳細了解可以參考:PROTAC —— 模塊化構建藥物分子的未來)。
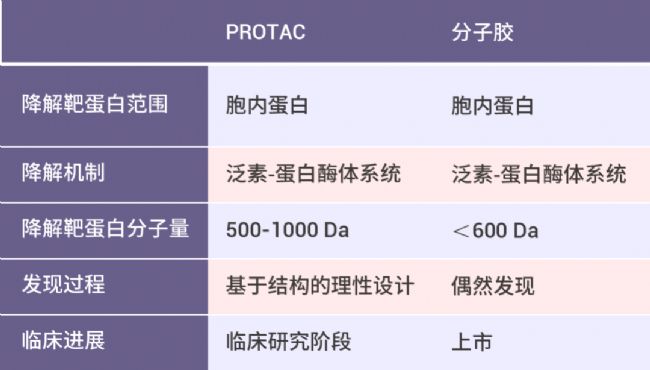
此外,除 PROTAC 外,分子膠也是基于泛素-蛋白酶體系統實現對靶蛋白的降解。由于分子膠產品分子量相對較小,具有更好的生物利用度。但目前報道的分子膠產品更多是來自于偶然發現,而 PROTAC 分子更容易通過理性設計進行開發。

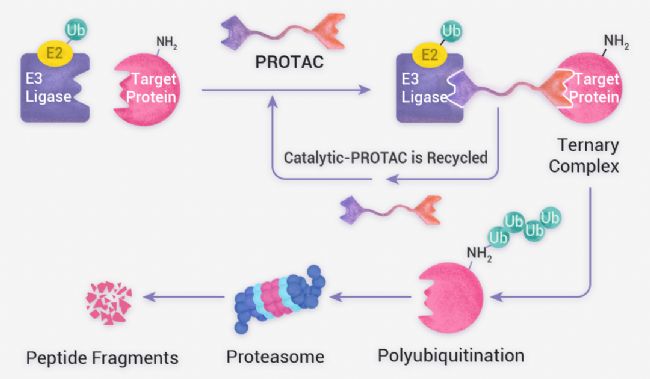
除了基于泛素‑蛋白酶體系統開發的雙功能分子,溶酶體靶向嵌合體 (LYTAC)、自噬靶向嵌合體 (AUTAC) 及 ATTEC 則是通過溶酶體途徑實現對靶蛋白的降解。不同雙功能分子具體作用機制及對靶蛋白的降解范圍不完全一樣,可以起到互補的作用 (如下表所示)。
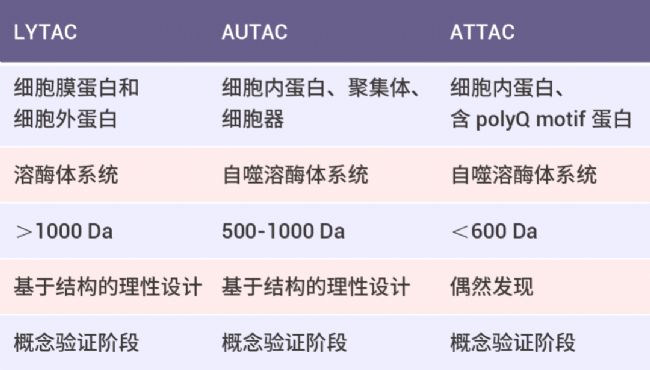
左右滑動查看更多
表 1. 不同雙功能分子比較[3]。

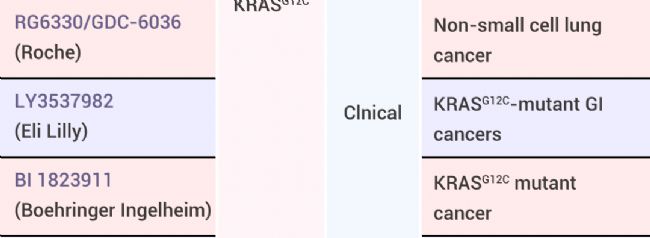
共價抑制劑一般通過溫和的反應性官能團以共價鍵結合到靶蛋白氨基酸殘基上,賦予額外的親和力。而非共價抑制劑,則是通過氫鍵或范德華力等非共價鍵相互作用,來實現對靶蛋白的結合和抑制。由于非共價相互作用相對較弱,為了保證非共價抑制劑的親和力,需要在靶蛋白表面形成較深結合口袋,使小分子能夠有效結合。然而,共價抑制劑可以針對缺乏表面“口袋”的“不可成藥”蛋白,可以擴大治療范圍。其中 KRASG12C 抑制劑 Sotorasib 的批準也是共價藥物發展和攻克“不可藥性”領域的一個重大里程碑。
KRAS 蛋白是一種無特征的、近球形的結構,沒有明顯的結合位點,很難合成能有效靶向和抑制其活性的化合物,且與內源性配體具有極高的結合活性,這也使得它不能像蛋白激酶抑制劑那樣產生有效的競爭。共價藥物的開發推動了 KRAS 藥物的發現。目前多款針對 KRAS 的共價抑制劑已經被開發進入臨床應用及研究,從而實現了 KRAS 從“不可成藥”到“可成藥”靶點的逆襲[4]。









目前開發的大多數靶向蛋白的調節劑,無論是激動劑、拮抗劑還是抑制劑,絕大多數都屬于正構類型 (Orthosteric) 的藥物分子,即藥物直接靶向受體的內源性分子結合點。然而,由于許多靶點與底物的親和力很高,缺乏結構信息或其活性位點高度保守,導致不可藥或難以在其正向位點上靶向。為了克服這些挑戰,變構調節被提出作為一種常用的策略。變構調節是通過配體在功能位點以外的部位結合,導致蛋白構象變化或蛋白質動力學變化,進而影響內源性配體與蛋白的結合。
變構位點的鑒定和相應的藥物設計為以前被認為在其正向位點上“不可藥性”或“難以靶向”的蛋白質開辟了新的治療機會。目前已經有多種變構藥物用于臨床實踐。

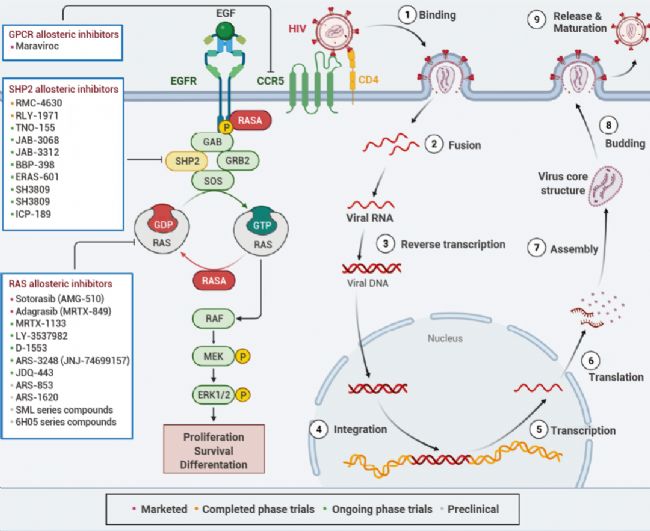
信號通路中已上市、臨床和臨床前共價抑制劑的圖譜。

蛋白互作 (PPIs) 參與機體多種生物進程,并通過形成蛋白質-蛋白質復合物或 PPI 網絡來發揮其功能。PPIs 的大而平坦的相互作用表面使得小分子調節劑的開發充滿挑戰。
目前開發的 PPIs 抑制劑主要包括小分子、抗體、多肽或重組蛋白等。但各存在優勢和不足,如小分子抑制劑親和力低,成藥性差,而多肽,抗體及重組蛋白雖然具有較高的結合活性,但存在穩定性低,生物利用度差等問題。為了改善多肽的穩定性,藥物研發人員也會采用環肽或擬肽來改善多肽藥物的成藥性。而針對抗體或重組蛋白藥物,高效遞送技術和小尺寸抗體模擬物 (如 scFv 和納米抗體) 的發展可以提高它們在治療不可成藥靶點中的應用。近年來,“熱點”位點的發現使得開發靶向 PPIs 藥物變得更加有效。熱點殘基是指在蛋白質之間形成結合位點的 PPIs 界面,可以通過識別熱點來設計靶向 PPIs 界面的藥物[5]。此外共價抑制和變構抑制也會用于 PPI 小分子抑制劑開發中。

基因治療是在基因或轉錄水平來調節基因表達的過程,突破了小分子藥物對靶蛋白位點的限制,大大擴展了藥物靶向的范圍。
目前 RNA 干擾技術、反義 DNA 技術、ASO 技術等前沿技術和方法的應用,為核酸藥物的研究和臨床應用提供了更為廣闊的發展空間,此外,CRISPR 基因組編輯也已經成為近年來最受矚目的科學突破之一。雖然基因治療仍面臨很多挑戰,但隨著技術的不斷發展和創新,基因治療將會帶來更多可能。

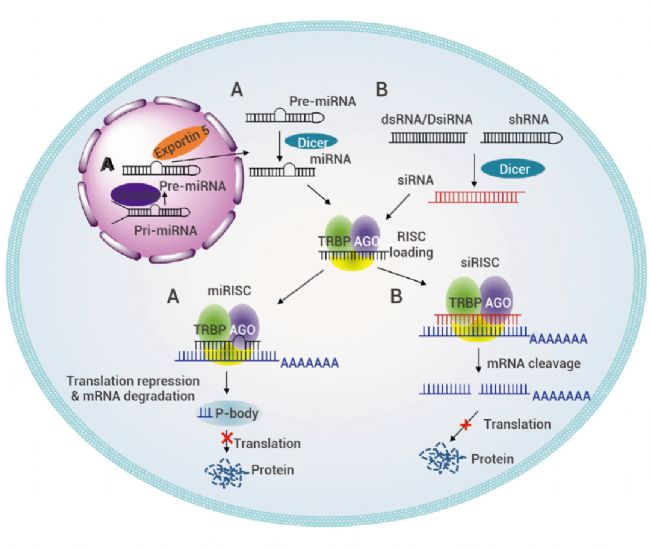
圖 4. miRNA (A )和 siRNA (B) 作用機制示意圖[6]。
目前開發的藥物靶點只是所有疾病靶點中的冰山一角,而未被開發的“不可成藥”靶點則是一片汪洋大海。雖然針對“不可成藥”靶點的藥物開發存在巨大挑戰,但隨著技術的不斷發展和完善,藥物靶點的范圍將進一步擴大,將有越來越多的“不可成藥”靶點被撕掉“不可成藥”標簽,將有更多的藥物被開發出來惠及人類健康。
 關于 MCE
關于 MCEMCE 助力藥物研發,針對“不可成藥”靶點,可以提供多種類型產品,包括 PROTAC 設計合成、寡核苷酸相關產品及定制合成、多種不同類型化合物庫,包括共價化合物庫、多肽庫、環肽庫、擬肽庫、蛋白互作化合物庫、大環化合物庫等,滿足針對“不可成藥”靶點藥物開發的不同需求。

[1] Xie X,et al. Recent advances in targeting the "undruggable" proteins: from drug discovery to clinical trials. Signal Transduct Target Ther. 2023 Sep 6;8(1):335.
[2] Lazo JS, et al. Drugging Undruggable Molecular Cancer Targets. Annu Rev Pharmacol Toxicol. 2016;56:23-40.
[3] Zhang G,et al. Strategies for targeting undruggable targets. Expert Opin Drug Discov. 2022 Jan;17(1):55-69.
[4] Huang L,et al. KRAS mutation: from undruggable to druggable in cancer. Signal Transduct Target Ther. 2021 Nov 15;6(1):386.
[5] Cukuroglu E,et al. Hot spots in protein-protein interfaces: towards drug discovery. Prog Biophys Mol Biol. 2014 Nov-Dec;116(2-3):165-73.
[6] Hu B,et al. Therapeutic siRNA: state of the art. Signal Transduct Target Ther. 2020 Jun 19;5(1):101.